G.T. RIJKERS 1, P.M.W. JANSSENS 2 en P. HERBRINK 3
Op 20 mei 1720 meerde het schip van kapitein Chataud met een
lading zijde en katoen uit Syrië af in de
haven van Marseille, destijds de stad met een monopolie voor
handel met het Midden-Oosten.
In Syrië heerste de pest en tijdens de reis waren 6
bemanningsleden overleden.
Kapitein Chataud had de lokale havenautoriteiten hiervan op de
hoogte gesteld, maar omdat zijn kostbare lading bestemd was voor
de grote jaarmarkt bij Arles mocht hij, op aandringen van
invloedrijke kooplieden, toch lossen.
Binnen enkele dagen brak de pest uit in Marseille. In een
periode van 2 jaar stierven 50.000 van de 90.000 inwoners van de
stad.
De angst dat deze plaag zich over de Provence zou verspreiden
was terecht groot. De doodstraf werd ingesteld voor ieder die
uit Marseille naar de Provence wilde vluchten. Als extra
verdediging bouwden de inwoners van Avignon en wijde omtrek ter
hoogte van Lagnes, halverwege Marseille en Avignon, de Mur de la
Peste (figuur 1), 50 km lang, 2 meter hoog en 70 cm dik.
Op geregelde plaatsen werden versterkte torens geplaatst met
uitkijkposten.
Deze eerstelijns verdediging tegen pathogene micro-organismen,
hoe goed bedoeld ook, heeft helaas
niet gewerkt. Ook in Avignon stierven 30.000 mensen aan de pest.
Deze geschiedenis illustreert hoe wankel het evenwicht tussen de
mens en zijn microbiële
omgeving is. Micro-organismen zijn altijd in de aanval en afweer
daartegen is permanent nodig.
Dit themanummer belicht de ‘chemie van de immunologie’.
Het geeft een overzicht van de diverse componenten van het
immuunsysteem. Onze inzichten in
de werking van het menselijk afweersysteem, en onze
mogelijkheden voor diagnostisch onderzoek hierin,
zijn de afgelopen halve eeuw exponentieel toegenomen.
Immunologie is thans een ver ontwikkeld vak met een duidelijk
afgebakend terrein en doel: het
immuunsysteem, respectievelijk de afweer en de hieraan
aanverwante processen waarmee het lichaam
reageert. De immunologie is anno 2004 een gedegen vakgebied waar
onderzoek wordt verricht op het gebied van cellen (lymfocyten,
fagocyten, granulocyten), moleculen (immunoglobulinen,
cytokinen, complement) en genen.
Het kennisterrein strekt zich uit in de breedte, maar gaat ook
in de diepte. De dominante
vraagstukken in de loop der tijd, en ongetwijfeld ook in de
toekomst, hebben te maken met moleculaire
biologie (DNA), celdifferentiatie en -regulatie en de
uitoefening van effectorfuncties die essentieel zijn
voor de afweer.
Dit imposante kennisterrein is het ijkpunt waartegen onderzoek
en toepassing van de immunologie
permanent worden afgemeten en een plaats gegeven.
Zoals professionals werkend met erfelijke stofwisselingsziekten
steeds teruggrijpen op het intermediaire metabolisme als
referentiepunt, zo keren professionals in de immunologie
(laboratoriumspecialisten en behandelaars) keer op keer terug
naar de basisimmunologie om te kunnen begrijpen waar ze mee
bezig zijn en om voorspellingen te kunnen doen.
Dat is ook zichtbaar in dit themanummer. Verschillende auteurs
beschrijven vanuit verschillende invalshoeken basisaspecten van
het immuunsysteem en bieden uitkijkjes naar kliniek en meer
specialistisch
laboratoriumonderzoek.
De term ‘immunologie’ wordt overigens op laboratoria soms ook
wel gebruikt om diagnostische tests aan te duiden waarbij
gebruik wordt gemaakt van immunologische principes (in een ELISA
bijvoorbeeld). Hieraan is welbeschouwd echter weinig
immunologie.
’Immunologie’ kan beter gereserveerd worden voor onderzoek dat
gerelateerd is aan ziekten van het immuunsysteem -een insteek
die aldus in dit themanummer is genomen.
Tegelijk met het toenemende inzicht in het functioneren van het
immuunsysteem heeft de medische
immunologie zich tot een herkenbaar specialisme ontwikkeld. In
de kliniek bestrijkt de immunologie
een breed spectrum van ziektebeelden, te vinden bij een groot
aantal specialismen. Het eerste oriënterendeonderzoek naar het
immuunsysteem zal bij patiënten in de dagelijkse praktijk worden
verricht op een klinisch-chemisch, medisch-microbiologisch of
immunologisch laboratorium.
Naast kennis van de immunologisch-diagnostische bepalingen is
hierbij ook inzicht in de mogelijkheden en indicatiestellingen
voor vervolgonderzoek van belang, zoals verschillende bijdragen
in dit themanummer illustreren.
Na een algemeen overzicht over de moleculen, cellen en weefsels
van het immuunsysteem (1), wordt ruim ingegaan op de diagnostiek
van haperende, aflatende of uit de hand gelopen afweer.
Vormen van tekortschietende afweer worden besproken in een
bijdrage over humorale immunodeficiënties (2).
Vervolgens worden de vroege processen tijdens de afweer
besproken, en dan met name de functie van complement hierin (3).
Bij monoklonale woekering van plasmacellen verschijnen
M-proteïnen in het bloed, wat ruimte biedt voor een evaluatie
van de kwaliteit van het onderzoek van deze eiwitten (4).
Een aparte categorie van ziektebeelden wordt gevormd door de
zogenaamde auto-inflammatoire ziekten. Deze worden in een
separate bijdrage besproken (5).
Voorts wordt aandacht besteed aan wat zou mogen worden
omschreven als manipulatie van de afweer.
Volgend op een algemeen overzicht over de structuur en functie
van immunoglobulinen, komen de vorming
van antistoffen na immunisatie en de in Nederland gebruikte
vaccinatieprogramma’s ruim aan bod (6).
Indien alle afweer ontbreekt is de infectiegevoeligheid zo hoog
dat correctie van het defect noodzakelijk
is. Onder dergelijke omstandigheden zouden de mogelijkheden van
stamceltransplantatie en gentherapie
en de daarmee geassocieerde problematiek aan de orde kunnen
komen (7).
De reumatologie en allergologie zijn de vakterreinen bij uitstek
waarin immunologie een rol speelt. Echter,
ook in de interne geneeskunde, longgeneeskunde,
gastro-enterologie, kindergeneeskunde, dermatologie
en neurologie passeren regelmatig immunologische vraagstukken.
In feite kan de clinicus in nagenoeg elk
medisch specialisme geconfronteerd worden met immunologische
problematiek. De voor diagnose, behandeling en beleid
noodzakelijke medische laboratoriumdiagnostiek valt onder de
discipline medische
immunologie, zoals die te vinden is in academische en grotere
regionale centra. Hier vindt men vaak ook
speciële medisch-immunologische laboratoria. Regelmatig zijn
daarnaast medisch immunologen werkzaam
binnen klinische-chemische of medisch-microbiologische/
immunologische laboratoria.
De speciële medisch-immunologische laboratoria komen met name in
beeld als de vraagstellingen dieper gaan of het zeldzamer
aandoeningen betreft. Daartoe moeten zij over een breed arsenaal
aan bepalingen beschikken en voortdurend inspelen op de nieuwste
inzichten en mogelijkheden.
De klinische toepassing van immunologische kennis is verre van
volledig. Toepassing wordt, vanzelfsprekend, bepaald door de
(financiële) mogelijkheden en is afhankelijk van de inzet en
inzichten van velen, niet alleen de immunologen. Een mooi
voorbeeld is vaccinatie -met afstand de meest kosteneffectieve
preventiemaatregel van de totale gezondheidszorg.
Het succes van de vaccinatie kan moeilijk (geheel) op het conto
van immunologen worden geschreven. Bij
de invoering van het Rijksvaccinatieprogramma waren namelijk nog
lang niet alle onderliggende immunologische principes bekend.
Zelfs de ‘evidence based medicine ’ stond nog in de
kinderschoenen. Tekortschietende antistofproductie wordt al
sinds 1952 behandeld met substitutie van gammaglobuline (8) -een
vorm van therapie die effectief is, maar wel belastend en niet
geheel zonder gevaren. Tekorten aan complementfactoren kunnen,
afgezien van C1-esteraseremmer, niet zonder meer worden
gesuppleerd, en middelen om de productie door het lichaam op te
laten voeren kennen we niet (een ‘search’ op PubMed met
als trefwoorden “complement” - “deficiency” - “treatment” en
“review” levert 0 hits op!).
Pas sinds kort wordt geïntervenieerd in ontregelde
cytokineresponsen (TNF, IL-1) bij reuma en andere inflammatoire
ziekten (ziekte van Crohn) (9, 10). De kosten van
deze vormen van behandeling zijn hoog en de langetermijneffecten
nog onbekend. Deze voorbeelden mogen duidelijk maken dat voor
implementatie van veel immunologische kennis in de kliniek de
echte oogstperiode nog moet starten.
De klinische betekenis van immunologie betreft trouwens lang
niet alleen de afweer tegen infecties en strijd tegen vreemde
indringers, zoals die in dit themanummer ruim aan bod komen.
Minstens even verstrekkend zijn de gevolgen van situaties
waarbij het afweersysteem verkeerd gericht is, zoals voorkomt
bij auto-immuunziekten, bijvoorbeeld reuma en SLE en vormen van
allergie en astma. Hieraan kan gemakkelijk een heel volgend
themanummer gewijd worden.
Het immuunsysteem is opgebouwd uit een groot aantal cellen en
moleculen, die door onderlinge interactie in de lymfoïde organen
zorgdragen voor een adequate immuunrespons bij infectie.
Het wordt onderverdeeld in aangeboren, niet specifieke
immuniteit en verworven, specifieke immuniteit.
Er is een aanzienlijke interactie tussen beide systemen.
Beide bevatten zowel humorale als cellulaire componenten.
Niet-specifieke immuniteit vormt, na niet-immunologische
factoren als huid en secreten, de eerstelijns verdediging tegen
infecties.
Tot de cellen van het niet-specifieke immuunsysteem behoren o.a.
granulocyten, monocyten, macrofagen, dendritische cellen en
NKcellen.
Naast cellulaire factoren zijn er ook een aantal humorale
factoren zoals complement en mannosebindend
lectine. Specifieke immuniteit komt wat langzamer op gang en
leidt tot geheugenvorming waardoor bij een tweede contact met
het micro-organisme een snelle reactie op gang komt.
De belangrijkste cellen van de specifieke immuniteit zijn de
T-cellen, die de cellulaire immuniteit verzorgen. Onder de
T-cellen hebben de CD4-positieve T-cellen een sterk regulerende
functie, waarbij cytokinen een belangrijke rol spelen. De
humorale immuniteit wordt verzorgd door B-cellen en hun
producten, de immunoglobulinen.
De mens leeft in een wereld omringd door micro-organismen:
virussen, bacteriën, schimmels en parasieten. Deze kunnen in 4
categorieën ingedeeld worden: onschuldige, goede, slechte
micro-organismen en extreem schadelijke ziekteverwekkers. Tot de
laatste categorie behoren micro organismen die ook in personen
met een intacte afweer kunnen leiden tot ernstige morbiditeit en
mortaliteit, zoals bij voorbeeld tetanus en ebola. Tot de
slechte categorie behoren de micro-organismen die in het
algemeen geen ziekte veroorzaken, maar in sommige gevallen wel
(bijvoorbeeld bij mensen met immuunstoornissen).
Van goede micro-organismen heeft de mens voordeel omdat zij
helpen bij spijsvertering, of door kolonisatieresistentie
voorkómen dat pathogene micro-organismen een infectie
veroorzaken. Onschuldige micro-organismen geven noch voordeel
noch nadeel voor de mens.
Het immuunsysteem heeft als taak om het lichaam te verdedigen
tegen infecties en mogelijk ook ongecontroleerde groei van
tumorcellen. Daar waar de afweer tekort schiet kan het
immuunsysteem worden versterkt door vaccinatie of door
immunotherapie. De effectormechanismen van het immuunsysteem
zijn destructief van aard: potentieel ziekteverwekkende
micro-organismen en tumorcellen moeten worden vernietigd. Dit
kan leiden tot aanzienlijke (immuno) pathologie. Het is derhalve
zaak het immuunsysteem onder controle te houden. Er dient een
balans te zijn tussen beschermende immuniteit en
immunopathologie cq. auto-immuniteit.
Cellen en moleculen van het immuunsysteem
Het menselijk lichaam wordt in de eerste plaats tegen
infecties beschermd door mechanische barrières zoals de huid
en slijmvliezen. Deze vormen niet alleen een fysieke, maar ook
een chemische barrière. Ook de normaal aanwezige microbiële
darmflora vormt een barrière. Door kolonisatieresistentie
wordt voorkomen dat pathogene micro-organismen een infectie
kunnen veroorzaken. Het immuunsysteem treedt pas in werking
wanneer deze barrières worden doorbroken.
Het immuunsysteem wordt onderverdeeld in aangeboren,
niet-specifieke immuniteit en verworven,
specifieke immuniteit. Beide soorten immuniteit kunnen niet
los van elkaar worden gezien, er is een
aanzienlijke interactie tussen beide systemen (1).
Niet-specifieke immuniteit
Niet-specifieke of aangeboren immuniteit (in de Angelsaksische
literatuur aangeduid als ‘innate immunity’) reageert snel op
het binnendringen van een micro-organisme (in een tijdsbestek
van minuten tot uren).
Tot de cellen van de niet-specifieke immuniteit behoren o.a.
granulocyten, monocyten, macrofagen en
dendritische cellen en NK-cellen. Neutrofiele granulocyten
spelen een belangrijke rol in de niet-specifieke
afweer. Dergelijke cellen bezitten het vermogen om na
adherentie aan geactiveerd vaatendotheel uit
de bloedbaan de treden (diapedese) en op geleide van een
concentratiegradiënt te migreren naar een ontstekingshaard,
een proces dat chemotaxie wordt genoemd.
Fagocyten bezitten op het celoppervlak receptoren voor
koolhydraten en lipopolysachariden
( Toll-like receptoren), waardoor ze rechtstreeks
micro-organismen kunnen binden en fagocyteren. Op deze wijze
wordt een snelle eerste afweerreactie gevormd.
De efficiëntie van binding en fagocytose wordt drastisch
verhoogd indien de micro-organismen beladen
zijn met antistoffen en complement. In het daaropvolgende
proces worden gefagocyteerde bacteriën gedood en verwijderd
door de vorming van zuurstofradicalen en enzymen in diverse
granulae (2). Defecten in chemotaxie, fagocytose of ‘killing’
kunnen leiden tot chronische of recidiverende infecties
(3).
Monocyten en macrofagen kunnen, evenals granulocyten,
micro-organismen binden, fagocyteren en doden.
Daarnaast kunnen ze evenals granulocyten biologisch actieve
stoffen uitscheiden. Anderzijds spelen macrofagen een rol in
de regulatie van de ontstekingsreactie en de activatie van de
specifieke immuniteit. Hierbij spelen gespecialiseerde
antigeen-presenterende cellen, met name dendritische cellen,
een belangrijke rol.
De cytotoxiciteit van natural-killer(NK)-cellen valt ook onder
niet-specifieke immuniteit. NK-cellen kunnen
tumorcellen en viraal geïnfecteerde cellen herkennen en doden,
al dan niet via Fc-receptoren die
celgebonden antistoffen kunnen binden. In het laatste geval
spreken we van antistof-afhankelijke cellulaire
cytotoxie (ADCC).
Naast de cellulaire factoren van de aspecifieke immuniteit
zijn er ook een aantal humorale factoren. Hiertoe behoren o.a.
complement, mannosebindend lectine (MBL) en stoffen als
lactoferrine en lysozym (2).
De aspecifieke afweer is weliswaar in staat om een infectie
enigszins in toom te houden, maar is vaak
niet voldoende om het micro-organisme volledig te
elimineren.
Specifieke immuniteit
Specifieke immuniteit komt trager op gang na een eerste
contact met een micro-organisme (in een
tijdsbestek van dagen, weken). Herhaald contact met hetzelfde
pathogeen leidt echter tot een sneller antwoord, dat bovendien
heviger is.
De specifieke immuniteit wordt opgedeeld in humorale en
cellulaire immuniteit (figuur 1).
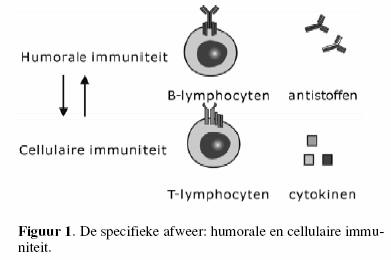
Onder humorale immuniteit verstaan we B-lymfocyten en hun
producten: de antistoffen, terwijl de cellulaire immuniteit
wordt verzorgd door T- lymfocyten en hun producten: cytokines.
Er bestaan 2 soorten Tlymfocyten: helper- T-lymfocyten (met
CD4-moleculen
aan het celoppervlak) en cytotoxische T- lymfo yten (met
CD8-moleculen aan het celoppervlak). Het herkenningsmolecuul
van B-lymfocyten is membraangebonden immunoglobuline. Via dit
molecuul wordt de B-lymfocyt specifiek geactiveerd door
antigeen -oplosbaar eiwit of polysaccharide, of celgebonden
antigeen.
De effectormoleculen van geactiveerde B-lymfocyten, de
antistoffen, hebben dezelfde specificiteit als de
membraangebonden antistof. Antigeenherkenning door
T-lymfocyten verloopt ook via specifieke receptoren
(T-celreceptor) met één verschil in vergelijking met
B-lymfocyten. De T-celreceptor herkent alleen antigenen die in
de vorm van peptiden gebonden aan HLA-moleculen worden
‘aangeboden’. Antigenen die door de cel zelf worden
geproduceerd (endogene antigenen) worden op een andere manier
gepresenteerd dan exogene antigenen zoals bacteriën. Endogene
antigenen, zoals virusantigenen en tumorantigenen, worden
gebonden in klasse-1-HLA-moleculen (HLA-A, -B en -C) (4).
Klasse-1-HLA-moleculen komen op alle kernhoudende cellen voor.
De T-celreceptor van een
CD8-cytotoxische T-lymfocyt bindt met antigenen in
klasse-1-HLA. Op deze manier kunnen cytotoxische Tcellen
virus-geïnfecteerde cellen en tumorcellen elimineren.
Exogene antigenen worden gepresenteerd gebonden in
klasse-2-HLA ( H L A - D P, -DQ en -DR).
C D 4 - T-lymfocyten herkennen antigenen gebonden in klasse -
2 - H L A (figuur 2).
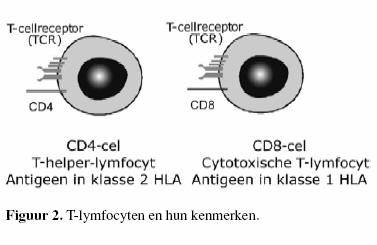
Klasse-2-HLA komt slechts op een beperkt aantal celtypen voor.
Antigeenpresentatie in klasse-2-HLA is derhalve voorbehouden
aan klasse - 2 - H L A positieve cellen, met name dendritische
cellen, monocyten, macrofagen en B-cellen.
CD4-T-helper-lymfocyten hebben vooral regulerende functies.
Het belang van HLA bij de cellulaire immuniteit wordt
geïllustreerd door patiënten met het zogenaamde ‘bare
lymphocyte’-syndroom. Bij deze patiënten ontbreekt
klasse-II-HLA (soms ook klasse I).
Daardoor is er een ernstige cellulaire immuundeficiëntie.
Op basis van hun regulatoire functie kunnen
T-helper-lymfocyten worden onderverdeeld in Th-1-, Th-2- en
Tr-cellen.
Th-1-cellen produceren met name IL-2 en interferon-gamma en
ondersteunen vooral de cellulaire immuunreactie.
Th-2-cellen produceren met name IL4 en IL5 en zijn meer
betrokken bij antistofproductie (5).
Het evenwicht tussen type-1- en type- 2-cytokinen is bij
infecties en ontstekingsprocessen vaak (tijdelijk) verstoord.
Bij een groot aantal tumoren is een abnormale productie van
IL-10 beschreven met als gevolg een verminderde cellulaire
immuunrespons (6). Tr-cellen produceren naast IL-10
ook‘transforming growth factor-β’ (TGF-β) en remmen de
Th-1-gemedieerde antitumorrespons (7).
Tegen veel micro-organismen wordt zowel een respons van
B-lymfocyten als van T-lymfocyten opgewekt.
Als vuistregel kan men echter zeggen dat de humorale
immuniteit (de B-lymfocyten en antistoffen) met
name anti-bacterieel werkt en de cellulaire immuniteit (
T-lymfocyten) met name anti-viraal of anti-tumor, alhoewel bij
lytische infecties ook neutraliserende antistoffen een
belangrijke rol spelen. Het specifieke immuunsysteem reageert
gericht op een binnendringend micro-organisme.
Zowel antistoffen als T-cel receptoren worden in een zodanige
verscheidenheid aangemaakt dat het repertoire in principe
volledig is, dit wil zeggen ieder mogelijk micro-organisme (of
onderdeel ervan, een antigeen-epitoop) wordt wel door een of
meerdere antistoffen of receptoren herkend.
Deze verscheidenheid komt tot stand in een proces van
herschikking van de genen tijdens de ontwikkeling van
B-lymfocyten in het beenmerg en van T-lymfocyten in de thymus.
In een volwassen individu omvat het humorale immuunsysteem
minimaal ongeveer 108 verschillende antistoffen (met
bijbehorende B-lymfocyten) en het cellulaire immuunsysteem
eveneens minimaal 108 verschillende T-lymfocyten (8).
Bij het eerste contact met een bepaald antigen wordt een
primaire reactie opgewekt. Dit eerste contact kan in de vorm
van een natuurlijk voorkomend micro-organisme zijn of in de
vorm van een vaccin. Hierdoor worden de B-lymfocyten
gestimuleerd die specifiek voor het antigen zijn. Deze
stimulatie of activatie maakt dat deze specifieke B-lymfocyten
zullen prolifereren (klonale expansie) en differentiëren tot
antistofproducerende plasmacellen of tot
geheugen-B-lymfocyten.
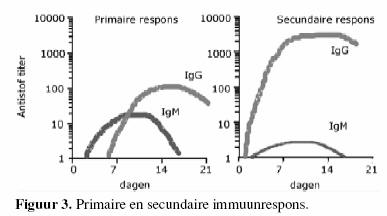
Deze reactie duurt 10-14 dagen. Bij een tweede contact met
hetzelfde antigen verloopt de activatie sneller (3-7 dagen) en
heviger. Indien een persoon na volledige vaccinatie in contact
komt met het natuurlijk voorkomend pathogeen zal een activatie
van geheugen-B-lymfocyten zeer snel leiden tot de aanmaak van
grote hoeveelheden neutraliserende antistoffen die de persoon
in kwestie zullen beschermen tegen ziekte (figuur 3). Voor de
vorming van antistoffen tegen de meeste antigenen zijn er
naast B-lymfocyten ook T- helpercellen nodig. De
antistofrespons op dit soort antigenen noemt men dan
T-cel-afhankelijk (figuur 4). Bacteriën met een
polysaccharidekapsel (bijvoorbeeld Haemophilus influenza type
b, pneumococcen, meningococcen) induceren een antistofrespons
zonder de hulp van
T-cellen.
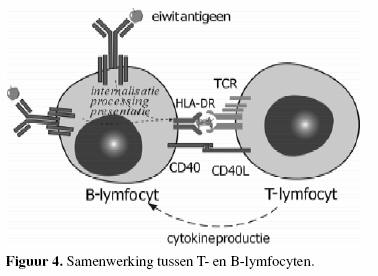
Daarom worden deze polysacchariden geklasseerd onder antigenen
die T-cel-onafhankelijk zijn (9).
Ook het T-cellulaire immuunsysteem ondergaat bij contact met
een antigeen klonale expansie en ontwikkelen zich bijkomend
geheugencellen (10).
Voor cytotoxische T-lymfocyten geldt dus evenals voor
B-lymfocyten dat voorafgaande vaccinatie het antwoord op een
latere natuurlijke infectie kan versnellen en versterken.
Opbouw en organisatie van het immuunsysteem
De aanmaak van lymfocyten vindt plaats in de primaire lymfoïde
organen: het beenmerg en de thymus.
In het beenmerg bevinden zich de pluripotente stamcellen.
Uit deze cellen ontstaan gecommitteerde (lymfoïde) stamcellen,
die kunnen differentiëren tot ofwel
voorloper- T-lymfocyt ofwel -B-lymfocyt.
Uitrijping van de voorloper- T-lymfocyt vindt in de thymus
plaats. Hier vindt het ingewikkelde proces van genherschikking
plaats waarbij door middel van recombinatie een grote
diversiteit aan T-celreceptoren ontstaat ( 11).
Omdat het proces ‘random’ geschiedt is er selectie nodig om de
T-cellen met een gewenste specificiteit
te behouden en de overigen uit te schakelen. Deze
selectieprocedure gaat in 2 fasen. Allereerst is er positieve
selectie waarbij de T-celreceptor moet kunnen binden met
HLA-moleculen op thymusepitheel. In
een tweede selectieronde worden de resterende Tlymfocyten die
lichaamseigen materiaal herkennen
uitgeschakeld (12).
Na dit selectieproces blijkt ongeveer 2-5% van de T-lymfocyten
aan de gestelde eisen te voldoen en komt als rijpe T-lymfocyt
in de circulatie.
De overige cellen sterven in de thymus door apoptose of worden
anderszins geïnactiveerd (13).
De uitrijping van B-lymfocyten vindt plaats in het beenmerg.
Tijdens dit proces wordt eveneens via genherschikking het
diverse repertoire van antigeenreceptoren van B-lymfocyten
(membraangebonden
immunoglobuline) gevormd (11).
Ook bij de vorming van B-lymfocyten kunnen cellen ontstaan met
ongewenste specificiteit voor lichaamseigen bestanddelen.
Deze cellen zijn in principe niet in staat tot auto-antistof
productie vanwege het ontbreken van specifieke
T-cel-help of andere tolerantiemechanismen.
Bovenstaand is beschreven hoe de cellen en moleculen van het
immuunsysteem lichaamsvreemde micro-organismen herkennen en
onschadelijk maken. Voorwaarde is wel dat de lymfocyt contact
maakt met het micro-organisme: ze moeten elkaar tegenkomen als
het ware. De lymfocyten bevinden zich vooral in lymfknopen, de
milt en gespecialiseerd lymfoïd weefsel langs de luchtwegen
(bronchus en ‘nasal associated lymphoid tissue’, BALT en NALT)
en de darm (‘gastrointestinal associated lymphoid tissue’,
GALT ) .
De circulatie van lymfocyten door het lichaam verloopt
geordend. Lymfocyten uit de bloedbaan dringen
lymfoïd weefsel binnen door de wand van postcapillaire
venulen. Via de lymfe worden ze weer afgevoerd
naar de ductus thoracicus en zo naar het bloed (14).
De ‘homing’ van lymfocyten op specifieke plaatsen
(bijvoorbeeld in GALT) wordt bepaald door receptoren op de
lymfocyt die kunnen binden aan tegenstructuren (adressinen) op
hoog-endotheel-venulen van het betreffend weefsel.
De zojuist beschreven organisatiegraad van het lymfoïde
weefsel is toch nog onvoldoende om adequaat
immunosurveillance te kunnen uitoefenen op alle plaatsen in
het lichaam. Immers, lymfocyten circuleren
niet door alle organen. Zo is er geen continue surveillance
van lymfocyten door de lever, bijvoorbeeld
om hepatitisvirus aldaar te herkennen. Voor dat doel bevatten
weefsels weefselmacrofagen (Kupffer-cellen in de lever,
Langerhans-cellen in de huid, etc.). Deze fagocyteren
lichaamsvreemd materiaal en transporteren dat naar drainerende
lymfknopen. Daar komt dan de specifieke respons van lymfocyten
op gang. Indien nodig worden de geactiveerde lymfocyten door
chemokinen naar de plaats van infectie getrokken (15, 16).
Initiatie van de immuunrespons
Zoals hierboven uiteengezet zijn er voor de mens als ‘good,
bad and ugly’ te beschouwen micro-organismen.
De afweer wordt niet bij ieder contact met een micro-organisme
geactiveerd; alleen wanneer er gevaar
zou kunnen ontstaan dient gereageerd te worden (17).
Activatie van de cellen van de niet-specifieke immuniteit is
hierin een belangrijke eerste stap. Deze activatie vindt
plaats door herkenning van pathogene structuren
(‘pathogen-associated molecular patterns’, PAMP ’s),
bijvoorbeeld LPS, peptidoglycanen, lipoproteïnen en bacterieel
CpG-DNA (18). Herkenning geschiedt door receptoren op de
cellen (‘pattern recognition receptor’, PRR), waarvan de ‘Toll
- like ’ receptoren (TLR) de laatste tijd erg in de
belangstelling staan. Tot op heden zijn er 10 TLR ’s
geïdentificeerd. TLR ’s komen tot expressie in zowel lymfoïd
als niet-lymfoïd weefsel, waarbij het expressiepatroon
varieert per celtype en weefsel (19).
Herkenning van pathogene structuren door T L R ’s leidt tot
activatie van de transcriptiefactor- NF-¥B en vervolgens tot productie van pro-inflammatoire cytokinen
als IL-1 en T N F -α.
In het geval van antigeen-presenterende cellen worden ook
co-stimulatoire moleculen tot expressie gebracht, bijvoorbeeld
B7-1 (CD80) en B7-2 (CD86) (20). Op deze wijze reguleert het
niet-specifieke immuunsysteem de expressie van belangrijke
co-stimulatoire moleculen en draagt bij aan het onderscheid
zelf/niet-zelf.

Ned Tijdschr Klin Chem Labgeneesk 2004; 29: 138-144
Humorale immuundeficiënties
M.J.D. van TOL 1, E.A.M. SANDERS 2 en G.T. RIJKERS 2
De laatste jaren is de kennis over de biologische oorzaken en
de klinische presentatie van de verschillende ziektebeelden,
waarin defecten in de productie van immunoglobulinen en van
antistoffen op de voorgrondtreden, enorm toegenomen.
Zo is van sommige ziektebeelden, zoals X-gebonden
agammaglobulinemie, het genetisch defect bekend en is voor
hyper-IgM-syndroom duidelijk geworden dat hieraan
verschillende genetische defecten ten grondslag kunnen liggen.
Ook voor ‘late onset’ hypogammaglobulinemie is recent een
eerste moleculair defect beschreven.
Teneinde artsen te ondersteunen bij het stellen van de
diagnose - belangrijk voor behandeling, preventie en
counseling - is een goede diagnostische aanpak en juiste
interpretatie van de laboratoriumbevindingen vereist.
In deze bijdrage wordt voor verschillende aangeboren humorale
immuundeficiënties een overzicht
gegeven van de immunologische afwijkingen, de genetische
defecten (indien bekend), de klinische
presentatie en de behandeling.
Immunoglobulinen zijn de effectormoleculen van de humorale
immuniteit. Voor een uitgebreide beschrijving van de
structuur, de vorming en de biologische eigenschappen van
immunoglobulinen, zie de bijdrage van Out et al., dit
themanummer (1).
Het inzicht in de biologische functie van de verschillende
immunoglobuline( sub)klassen is aanzienlijk vergroot door het
bestuderen van patiënten bij wie één, meerdere, of alle
(sub)klassen van immunoglobulinen afwezig zijn.
Deze zogenaamde humorale immuundeficiënties worden ook wel
‘experimenten van de natuur’ genoemd, omdat de klinische
symptomatologie van betrokken patiënten veel kan leren over de
specifieke functies van componenten van de humorale afweer.
Kennis van de normale fysiologie en de variatie daarbinnen is
onontbeerlijk voor correcte interpretatie van de pathologie,
in dit geval van humorale immuundeficiënties.
Het is normaal dat een pasgeborene start terwijl de
concentratie van IgG, dat via de placenta
van de moeder wordt verkregen, bij een voldragen zwangerschap
gelijk is aan die van de moeder.
Het maternale IgG verdwijnt met een halfwaardetijd van 21
dagen; tegelijkertijd neemt het vermogen van het kind om zelf
IgG te produceren toe.
Op de zuigelingenleeftijd is de periode tussen de 6e en de 18e
maand kritisch: voorbijgaande hypogammaglobulinemie van de
zuigeling treedt op wanneer het maternale mIgG verbruikt is en
de eigen IgG-productie nog niet goed op gang is gekomen.
Een juiste interpretatie van de humorale immuunstatus van
kinderen vereist daarom de beschikbaarheid en toepassing van
correcte leeftijdsafhankelijke normaalwaarden voor de
immunoglobulineklassen en IgG-subklassen.
De in Nederland gehanteerde leeftijdsafhankelijke
normaalwaarden zijn weergegeven in tabel 1 (2).

Het klinische spectrum van humorale immuundeficiënties omvat
ernstige fenotypes zoals bij X-gebonden
agammaglobulinemie, maar ook milde fenotypes zoals bij
IgA-deficiëntie, de meest voorkomende
vorm van humorale immuundeficiëntie.
Over het algemeen worden humorale immuundeficiënties
gekenmerkt door recidiverende, soms ernstige verlopende,
meestal bacteriële, infecties.
De meest frequent optredende deficiënties betreffen relatief
milde stoornissen zoals IgA-deficiëntie,
IgG-subklassedeficiëntie en
anti-polysaccharide-antistofdeficiëntie, of combinaties
daarvan.
De ernstige vormen van deficiënties, zoals
hypogammaglobulinemie met of zonder hyper-IgM- of
agammaglobulinemie zijn zeldzaam en worden daardoor slechts
zelden gezien.
Adequate diagnostiek is evenwel belangrijk, zodat tijdig de
juiste behandeling kan worden ingesteld ter voorkoming van
blijvende secundaire schade door de verhoogde
infectiefrequentie.
In deze bijdrage zullen de verschillende humorale
immuundeficiënties kort worden besproken.
Hierbij zullen het genetisch defect, de biologische
consequenties, de klinische presentatie en behandeling, en de
laboratoriumdiagnostiek worden belicht.
Het volledige diagnostische traject voor patiënten met (een
verdenking van) een humorale immuundeficiëntie omvat
serologisch, cellulair en moleculair onderzoek.
Het merendeel van de bekende moleculaire oorzaken van humorale
immuundeficiënties zijn intrinsieke defecten van de
B-lymfocyt.
T-lymfocytdefecten kunnen echter ook leiden tot een humorale
immuundeficiëntie
(zie hyper-IgM-syndroom).
Voor een overzicht van primaire immuundeficiënties in het
algemeen wordt verwezen naar de meest recente versie van een
regelmatig bijgesteld rapport van de IUIS (3).
In een recent overzicht van Ballow worden de humorale
immuundeficiënties uitstekend samengevat (4).
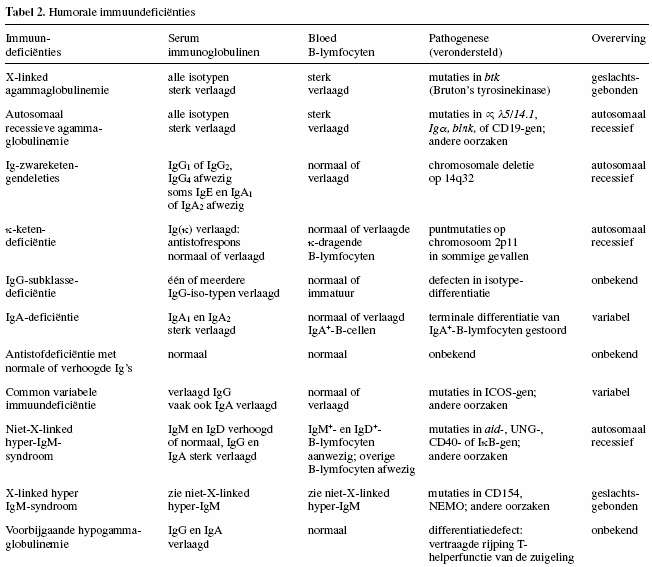
Tabel 2 geeft een overzicht van de moleculaire defecten, de
wijze van overerving en de
algemene bevindingen betreffende de serumimmunoglobulinen en
de aantallen B-lymfocyten in de circulatie bij de
verschillende ziektebeelden.
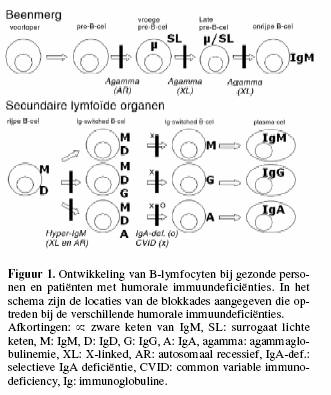
Figuur 1 representeert een algemeen schema voor de
B-celontwikkeling, waarin de blokkades zijn aangegeven die
optreden bij verschillende humorale immuundeficiënties.
IgA-deficiëntie
IgA-deficiëntie is de meest voorkomende immuundeficiëntie:
1:400 à 1:800 personen van Kaukasische
etnische origine is aangedaan (5, 6).
Er wordt van selectieve IgA-deficiëntie gesproken als de
IgA-waarde in serum onder 0,05 g/l ligt en de concentraties
van de overige immunoglobulinen (sub)klassen voor de leeftijd
normaal zijn.
Omdat de concentratie van IgA in serum bij pasgeborenen nog
zeer laag is, en slechts rond de puberteit volwassen waarden
worden bereikt, is het op de kinderleeftijd gebruikelijk als
criterium voor IgA deficiëntie een ondergrens voor IgA van het
gemiddelde minus tweemaal de standaarddeviatie
voor de leeftijd aan te houden.
Bedenk hierbij dat bij jonge kinderen tot de leeftijd van 4
maanden een IgAwaarde beneden de detectiegrens van
conventionele nefelometrie nog ‘normaal’ kan zijn (zie tabel
1).
In het algemeen is daarom beneden de leeftijd van 1 jaar een
IgA-deficiëntie niet of zeer moeilijk vast te stellen.
IgA-deficiëntie komt meestal sporadisch, maar ook familiair,
voor. De wijze van overerving, autosomaal
dominant of autosomaal recessief, is echter onbekend.
IgA-deficiëntie is tevens beschreven in families waarin ook
een andere vorm van primaire
immuundeficiëntie, namelijk ‘common variable immunodeficiency’
(CVID), voorkomt.
Sporadisch zijn deficiënties van één van de twee IgAsubklassen
beschreven. In deze gevallen wordt het
defect meestal veroorzaakt door een deletie van het gen dat
codeert voor de betreffende subklasse. In verreweg de meeste
gevallen is echter de oorzaak voor IgA-deficiëntie onbekend.
In de circulatie worden
normale aantallen B-lymfocyten aangetroffen, waarbij ook
B-cellen met IgA op het celmembraan kunnen
worden gedetecteerd, maar IgA-bevattende plasmacellen in het
beenmerg zijn sterk verlaagd of afwezig.
De differentiatie van mature B-lymfocyten, die IgA op het
membraan tot expressie brengen, naar IgA-producerende
plasmacellen lijkt dus gestoord.
Theoretisch kan dit veroorzaakt worden door een intrinsiek
defect in B-lymfocyten of door een defect op het niveau van
T-lymfocyten.
Het is belangrijk om zich te realiseren dat IgA-deficiënties,
soms in combinatie met IgGsubklassedeficiënties, secundair
kunnen optreden na congenitale infecties (rubella,
cytomegalovirus en Toxoplasma gondii), en ook geïnduceerd
kunnen worden door medicatie voorgeschreven bij epilepsie en
auto-immuunziekten (met name reumatoïde artritis).
IgA is vooral van belang als component van externe secreties
(speeksel, tranen, mucuslaag op respiratoir
en gastro-intestinaal epitheel).
Een selectieve IgA-deficiëntie heeft meestal geen klinische
consequenties.
Wellicht heeft dit te maken met het feit dat secretoir IgM
gedeeltelijk de rol van secretoir IgA kan overnemen.
Indien wel klinische consequenties aanwezig zijn, worden naast
recidiverende bacteriële infecties
van de luchtwegen en het KNO-gebied, met name moeilijk
behandelbare gastro-intestinale infecties met
Giardia lamblia gezien.
IgA-deficiëntie kan gecombineerd zijn met
IgG-subklassedeficiëntie en/of antipolysaccharide-
antistofdeficiëntie.
Bij circa 20% van de personen met IgA-deficiëntie wordt ook
een IgGsubklassedeficiëntie, meestal van IgG2 soms in
combinatie met IgG4, gevonden.
Onderzoek bij kinderen met IgA-deficiëntie én recidiverende
luchtweg- en KNO-infecties toonde bij circa 40% ook een
antipolysaccharide-antistofdeficiëntie aan.
Deze bevindingen onderstrepen het belang van bepaling van
zowel concentraties van immunoglobulinen als van specifieke
antistoffen bij verdenking van een humorale
immuundeficiëntie.
Daarnaast zijn associaties beschreven van IgA-deficiëntie met
allergie, auto-immuunziekten (o.a. van
het maagdarmkanaal), coeliakie, maligniteiten, en zelfs
mentale retardatie. Men moet er op bedacht zijn
dat de serologische diagnostiek van coeliakie met name berust
op detectie van IgA-antistoffen gericht
tegen ‘tissue transglutaminase’ (tTG), die bij een complete
IgA-deficiëntie uiteraard ontbreken. Alleen
histologisch onderzoek van vlokatrofie in een dunnedarmbiopt
kan dan uitsluitsel geven.
Bij patiënten met een complete IgA-deficiëntie kunnen bij
transfusie van IgA-bevattende bloedproducten
of immunoglobulinepreparaten anti-IgA-antistoffen gevormd
worden. Deze antistoffen kunnen bij herhaalde expositie aan
IgA een anafylactische reactie uitlokken.
Bij het toedienen van een bloedtransfusie aan een willekeurig
persoon wordt met het optreden
van deze reactie echter geen rekening gehouden, tenzij iemand
bekend is met een complete IgA-deficiëntie of zich bij een
persoon eerder problemen hebben voorgedaan.
Behandeling van patiënten met IgA-deficiëntie bestaat
voornamelijk uit het zo nodig profylactisch
toedienen van antibiotica bij patiënten met een verhoogde
incidentie van infecties of met ernstig verlopende infecties,
en/of uit een zorgvuldig bewaken van het optreden van
infecties en tijdig ingrijpen middels behandeling met
antibiotica.
IgG-subklassedeficiëntie
Het totale serum-IgG omvat vier subklassen met de
tussenhaakjes aangegeven verdeling: IgG1 (60-70%),
IgG2 (20-30%), IgG3 (5-10%) en IgG4 (1-5%) (zie ook tabel 1).
Bij een normaal totaal serum-IgG-gehalte kan er dus toch
sprake zijn van een deficiëntie van één of meer van de
IgG-subklassen.
De aanwezigheid van een IgG-subklassedeficiëntie hoeft,
evenals aangegeven voor IgA-deficiëntie, overigens niet per
definitie klinische betekenis te hebben.
Zoals genoemd bij IgA-deficiëntie, wordt ook een
IgG-subklassedeficiëntie gedefinieerd als de aanwezigheid van
een subklasse in een concentratie lager dan het gemiddelde
minus tweemaal de standaarddeviatie voor gezonde personen van
dezelfde leeftijd als de patiënt.
Het vermogen om de verschillende IgG-subklassen te produceren
vertoont een uiteenlopende
leeftijdsafhankelijkheid. Globaal gesproken is een kind
beneden de leeftijd van twee jaar relatief veel
beter in staat IgG1 en IgG3 te produceren dan IgG2 en IgG4.
Dit onderstreept opnieuw het belang van het toepassen van
leeftijdsafhankelijke normaalwaarden
voor het correct interpreteren van de kwantitatieve resultaten
van IgG-subklassen bij kinderen.
Slechts in sporadische gevallen wordt een selectieve
deficiëntie van één of meerdere IgG-subklassen
veroorzaakt door een deletie van het gen ( de genen ) coderend
voor het (de) betreffende IgG-subklasse(n).
In zijn algemeenheid wordt verondersteld dat
IgGsubklassedeficiënties het gevolg zijn van een afwijkende
regulatie van de expressie van de genen die coderen voor de
zware ketens van de IgG-subklassen
(7).
Recidiverende infecties van de bovenste en onderste
luchtwegen, met name veroorzaakt door gekapselde bacteriën
zoals Streptococcus pneumoniae komen bij patiënten met
IgG-subklassedeficiëntie (met name IgG2) vaker voor dan in de
algemene populatie.
Levensbedreigende bacteriële infecties met ernstige
complicaties en restverschijnselen zoals bronchiëctasieën zijn
bij geïsoleerde IgG-subklassedeficiëntie echter een zeldzaam
verschijnsel.
Antistoffen tegen eiwitantigenen, en een gedeelte van de
anti-polysaccharideantistoffen, zijn van de IgG1-
subklasse.
Een IgG1-subklassedeficiëntie resulteert (vaak) in een
verlaagd totaal serum-IgG, en dus in
hypogammaglobulinemie. Dit leidt altijd tot klinische
consequenties (zie verder).
Op de kinderleeftijd is een verlaagd IgG2 de meest voorkomende
IgG-subklassedeficiëntie.
De IgG2-subklasse is, naast de IgG1-subklasse, belangrijk voor
de antistofrespons op polysacchariden. Vandaar dat een
IgG2-subklassedeficiëntie klinisch vaak geassocieerd is met
een verhoogde infectiegevoeligheid voor bacteriën met een
polysaccharidekapsel, zoals Strepto - coccen.
Antistoffen tegen eiwitantigenen kunnen ook van de
IgG3-subklasse zijn. Vandaar dat IgG3-subklassedeficiëntie in
het algemeen wel gepaard gaat met klinische verschijnselen in
de vorm van een verhoogde infectiegevoeligheid.
IgG3-subklassedeficiënties worden vaker bij volwassenen dan
bij kinderen gezien.
Antistoffen tegen parasieten kunnen van de IgG4-subklasse
zijn. In sommige studies wordt wel een
associatie tussen IgG4-subklassedeficiëntie en recidiverende
luchtweginfecties beschreven, maar over het algemeen heeft een
solitaire IgG4-subklassedeficiëntie geen klinische
betekenis.
Patiënten met een IgG-subklassedeficiëntie en een bewezen
verhoogd risico op infecties worden vaak
profylactisch behandeld met antibiotica. Ook kan bij deze
patiënten intraveneuze of subcutane toediening
van immunoglobulinen overwogen worden.
Anti-polysaccharideantistofdeficiëntie
Een jong kind is in het eerste levensjaar al goed in staat
antistoffen tegen eiwitantigenen zoals difterie
en tetanus te maken. De productie van antistoffen tegen
polysaccharide-antigenen komt daarentegen pas vanaf het tweede
en derde levensjaar echt op gang, en is pas volledig
uitgerijpt aan het einde van de eerste decade.
Als mogelijke oorzaken voor het vertraagd uitrijpen van het
vermogen om op polysaccharideantigenen
te responderen zijn zowel een onrijpheid van de B-lymfocyten
als van antigeenpresenterende
cellen in de milt gesuggereerd.
Het blijkt dat bij 15-25% van de kinderen met recidiverende
bacteriële luchtweginfecties een verlaagde
(<20% van normaal) anti-polysaccharideantistofrespons wordt
gevonden na vaccinatie met niet-geconjugeerde
polysaccharideantigenen van Haemophilus of pneumococcen,
ondanks de aanwezigheid van normale concentraties van de
immunoglobulinenklassen en IgG-subklassen in het bloed en een
normale antistofrespons na vaccinatie met eiwitantigenen.
Deze deficiëntie wordt in de literatuur ook wel omschreven met
de termen “selectieve antilichaamdeficiëntie” of
“antigeen-specifieke antilichaamdeficiëntie”.
Bovendien wordt bij kinderen met recidiverende
luchtweginfecties en een selectieve of gecombineerde IgA en/
of IgG2-subklassedeficiëntie zelfs in 40% van de gevallen een
geassocieerde anti-polysaccharideantistofdeficiëntie gevonden.
Een anti-polysaccharideantistofdeficiëntie resulteert
doorgaans in klinische verschijnselen, die echter sterk kunnen
variëren in ernst: geen problemen, recidiverende bacteriële
bovenste luchtweginfecties,
recidiverende pneumonieën met bronchiëctasieën, en
levensbedreigende invasieve
infecties zoals meningitis. Dit laatste wordt met name gezien
als de antistofrespons op meerdere
pneumococcen-serotypen lager is dan 10% van normaal.
Evenals bij IgA- en/of IgG-subklassedeficiënties worden
profylactisch antibiotica gegeven, indien klinisch
vereist. Als dit onvoldoende resultaat heeft, kan intraveneuze
toediening van immunoglobulinen worden
overwogen (8, 9).
Overigens illustreren patiënten met een
anti-polysaccharide-antistofdeficiëntie zeer duidelijk dat
kwantificering van IgG-subklassen als zodanig slechts van
beperkte diagnostische betekenis is. Immers, bij deze
patiënten is sprake van een humoraal defect zonder dat één of
meerdere van de (sub)klassen van de immunoglobulinen
significant verlaagd zijn.
Agammaglobulinemie
De eerste stappen van B-lymfocytendifferentiatie vinden plaats
in het beenmerg, waar hematopoietische
stamcellen zich ontwikkelen tot voorloper-B-lymfocyten (figuur
1).
Voor de ontwikkeling tot B-lymfocyt is op DNA niveau
herschikking nodig van V(ariable ) - , D(iversity)-, en
J(oining)-gensegmenten en genen coderend voor het constante
gedeelte van de zware
(heavy, H) en lichte (light, L) ketens van
immuunglobulinen.
De recombinatie van deze V(D)J-genproducten met de zware keten
van IgM (µ-keten) en de expressie van IgM op het celmembraan
vormen belangrijke ‘checkpoints’ in de vroege ontwikkeling
van
B-lymfocyten. In het geval van een rijpingsstop die vroeg in
de B-celontwikkeling ligt, zullen rijpe Blymfocyten totaal of
nagenoeg volledig ontbreken in het perifere bloed, en worden
weinig tot geen immunoglobulinen gevormd.
Secundaire lymfoïde organen (lymfklieren, tonsillen) zijn
nauwelijks traceerbaar en plasmacellen in het beenmerg worden
slechts sporadisch aangetroffen. Er is dan sprake van
agammaglobulinemie
(IgG < 2 g/l, IgM < 0,2 g/l en IgA <0,05 g/l).
Humorale immuundeficiënties waarbij een selectieve blokkade
optreedt in de ontwikkeling van B-lymfocyten in het beenmerg
komen voor met een zeer lage frequentie van 1:200.000.
Deze deficiënties worden gekenmerkt door ‘early onset’
hypogammaglobulinemie en een sterk verlaagd, soms nagenoeg
volledig afwezig, aantal B-cellen in het bloed.
Belangrijk is hierbij te bedenken dat transplacentair
verkregen maternaal IgG in de eerste levensmaanden de
zuigeling beschermt, maar ook de aandoening bij
laboratoriumonderzoek in de eerste maanden maskeert.
Ongeveer 85% van patiënten met dit fenotype zijn jongetjes met
X-gebonden agammaglobulinemie
(XLA).
XLA is de langst bekende immuundeficiëntie en voor het eerst
beschreven in 1952 door Bruton
(10).
XLA wordt gekenmerkt door ernstige en recidiverende,
voornamelijk bacteriële, infecties. Het
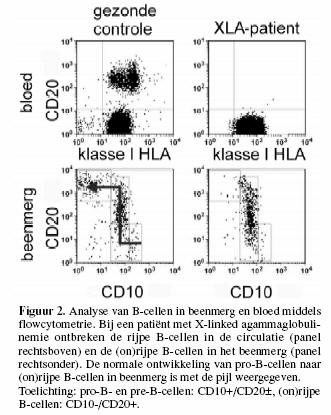
aantal rijpe B-lymfocyten in de circulatie is sterk verlaagd
evenals de concentraties van alle immunoglobulinen(sub)klassen
in serum (figuren 2 en 3).
Het aantal vroege voorloper-B-lymfocyten in het beenmerg,
pro-B-cellen of vroege pre-B-cellen, is normaal, maar ook in
het beenmerg zijn de aantallen cellen in de latere
ontwikkelingsstadia, namelijk late
pre-B-cellen (afhankelijk van het exacte defect), onrijpe en
rijpe B-cellen, sterk verlaagd of zelfs afwezig
(figuur 2).

In 1993 is ontdekt dat XLA optreedt ten gevolge van mutaties,
inserties en deleties in het gen dat codeert voor het enzym
Bruton’s tyrosine-kinase (btk).
De precieze functionele rol van btk in de ontwikkeling van
vroege stadia van de B-cellijn tot rijpe
B-lymfocyten is nog niet duidelijk.
Niet bij alle jongetjes met de klinische en
laboratoriumkenmerken van XLA wordt een defect in het
Btk-gen gevonden. Bovendien is 5-10 % van de papatiënten met
‘early-onset’ agammaglobulinemie en afwezigheid mvan B-cellen
een meisje.
Deze observaties suggereren dat er ook autosomaal recessieve
ziekten zijn met een fenotype dat grote overeenkomsten
mvertoont met XLA.
Inderdaad zijn recent vijf verschillende defecten beschreven
waarbij genen zijn aangedaan die coderen voor eiwitten,
respectievelijk de zware keten van IgM (µ-keten), BLNK,
Ig (CD79a), 5/14.1 en LRRC8, die een rol
spelen bij de ontwikkeling van voorloper B-cellen naar onrijpe
B-lymfocyten in het beenmerg (11).
Infecties van de luchtwegen treden vanaf de tweede helft van
het eerste levensjaar op de voorgrond, na
verdwijnen van maternale IgG-antistoffen, en verder worden
meer frequent huidinfecties, gastro-enteritis,
artritis en meningo-encefalitis bij patiënten met
agammaglobulinemie gezien. Haemophilus influenza
en Streptococcus pneumoniae worden frequent gediagnosticeerd
als ziekteverwekkers.
De T-cel-gemedieerde immuniteit is intact en daardoor worden
virale en schimmelinfecties zelden aangetroffen.
Een uitzondering hierop vormen chronische infecties met
enterovirussen, waaronder ECHO-virus dat een bekende oorzaak
is van virale meningo-encefalitis in deze groep van
patiënten.
Levenslange substitutie met immunoglobulinenpreparaten (IVIG,
intraveneus immunoglobuline, of subcutaan toegediend
immunoglobuline) kan de verhoogde gevoeligheid voor infecties
van zowel de lagere luchtwegen, van het KNO-gebied, als ook de
encefalitis grotendeels ondervangen.
Waakzaamheid op het optreden van infecties is echter geboden
en adequate behandeling van infecties met antibiotica vormt
een belangrijk onderdeel van de zorg voor patiënten met
agammaglobulinemie. Darminfecties met Giardia lamblia zijn
evenwel moeilijker te bestrijden, omdat slijmvliesgebonden
immuunglobuline (IgA) ontbreekt.
Meningo-encefalitis veroorzaakt door enterovirussen
(ECHO-virus) is een specifiek en ernstig
probleem van deze patiëntengroep, en laat vrijwel altijd een
fatale afloop zien. Dit treedt vooral op bij
patiënten die onvoldoende met immunoglobulinen worden
gesubstitueerd. Gentherapie voor btk bij patiënten met XLA
lijkt vooralsnog geen reële optie voor behandeling, omdat het
waarschijnlijk essentieel
is dat de expressie van het btk-gen onder een strikte controle
staat ter voorkoming van maligniteiten.
Bij een groot deel van de patiënten met agammaglobulinemie is
de genetische oorzaak van de ziekte vastte stellen. Dit
betekent dat in veel families erfelijkheidsadviesen prenatale
diagnostiek mogelijk is.
Hypogammaglobulinemie met hyper-IgM
Bij een hypogammaglobulinemie met normale of in sommige
gevallen verhoogde concentraties van IgM
en normale aantallen B-lymfocyten in het bloed kan sprake zijn
van het hyper-IgM-syndroom.
Hiervan zijn verschillende vormen te onderscheiden, de twee
geslachtsgebonden vormen (HIGM1- en NEMO-defecten) en diverse
niet-geslachtsgebonden vormen.
In het algemeen is ongeveer 60% van de patiënten bij wie
uiteindelijk de diagnose hyper-IgM wordt gesteld een jongetje.
HIGM1 wordt veroorzaakt door de afwezigheid van een
functioneel CD40-ligand (CD154) op geactiveerde
CD4+-T-lymfocyten en niet door een intrinsiek defect in de
B-cellen (12).
Strikt genomen is hier dus sprake van een cellulaire en niet
van een humorale immuundeficiëntie.
Zowel mutaties, inserties en deleties in het CD154-gen zijn
beschreven.
Bij sommige patiënten komt CD154 wel op het membraan van
geactiveerde T-cellen tot expressie, maar is het molecuul niet
functioneel en is de interactie met CD40 gestoord.
B-lymfocyten zijn normaal aanwezig in het perifere bloed. Deze
B-lymfocyten zijn, bij stimulatie door antigeen in de
secundaire lymfoïde organenin aanwezigheid van T-lymfocyten,
niet in staat over te schakelen van de productie van IgM
(en IgD) naar de productie van IgA, IgG en IgE.
Hiervoor is namelijk een interactie essentieel tussen
CD40-ligand op de geactiveerde T-lymfocyt en CD40 op de
B-lymfocyt.
Naast recidiverende infecties van de luchtwegen en het
KNO-gebied met gekapselde
bacteriën, treden bij deze patiënten ook opportunistische
infecties zoals Pneumocystis carinii-pneumonie, en chronische
diarree met progressief leverfalen door Cryptosporidium op.
De reden hiervoor is dat ook de interactie tussen geactiveerde
T-cellen en macrofagen
die CD40 tot expressie brengen gestoord is, waardoor activatie
van de macrofaag en het doden van intracellulaire
micro-organismen niet efficiënt optreedt.
Autosomaal recessief overervend hyper-IgM-syndroom kan worden
veroorzaakt door een mutatie in het ‘activation induced
cytidine deaminase’(AID)-gen (HIGM2).
AID-eiwit komt specifiek tot expressie in B-lymfocyten in de
secundaire lymfoïde organen (m.n.
lymfeklieren) die betrokken zijn bij een actieve
immuunrespons.
Dit eiwit is eveneens noodzakelijk voor de zogenaamde
immunoglobulineklasseswitch: het overschakelen van de
productie van IgM naar de productie van IgA, IgG en IgE.
Andere oorzaken van autosomaal hyper-IgM zijn defecten in CD40
(HIGM3) en het enzym uracil-DNA-glycosylase.
Hyper- I g M -syndromen kunnen ook veroorzaakt worden door
defecten in verschillende moleculen, zoals NEMO (Xgebonden
hyper-IgM) en I-B (autosomaal hyper- I g M ) die
betrokken zijn bij de regulatie van de translocatie van
NF-B van het cytoplasma naar de celkern (13).
Het (immunologische) fenotype van de hierboven beschreven
moleculaire defecten die aanleiding kunnen
geven tot hyper-IgM-syndroom kan sterk uiteen lopen.
Hieronder wordt verder alleen ingegaan op HIGM1.
Bij diagnose is de concentratie van IgM in het serumnormaal of
verhoogd, en zijn de concentraties van
IgG en IgA sterk verlaagd of zelfs niet detecteerbaar.
Bij vaccinatie met een ‘recall’-antigeen zoals tetanustoxoïd,
wordt geen secundaire antistofrespons geïnduceerd:
er is geen immunoglobulineklasseswitch van IgM naar IgG en
IgA, geen aviditeitsrijping van de antistofrespons en er
worden geen geheugen-B-cellen geïnduceerd.
CD4+- en CD8+-T-cellen zijn in normale aantallen in de
circulatie aanwezig. De in-vitro proliferatieve-respons
van T-cellen op mitogenen is normaal, maar de respons op
‘recall’ antigenen is verlaagd.
De oorzaak hiervoor is waarschijnlijk dat invivo-priming’ van
T-cellen door CD40-positieve
antigeenpresenterende cellen niet goed verloopt.
Om de diagnose met zekerheid te kunnen stellen is
mutatieanalyse van het CD154-gen essentieel.
Behandeling bestaat uit suppletie van de immuunglobulinen
middels IVIG en profylactische behandeling
met antibiotica ter preventie van Pneumocystis
cariniipneumonie. Ondanks deze maatregelen is de overleving op
40-jarige leeftijd slechts 30% vanwege het optreden van
leverfalen na Cryptosporidium-infecties of fataal verlopende
infecties als Pneumocystis carinii.
Stamceltransplantatie met een HLA-identieke donor is de beste
optie, eventueel zelfs gevolgd door levertransplantatie.
Gentherapie is waarschijnlijk geen optie voor behandeling,
omdat de expressie van CD154
sterk gereguleerd is en moet zijn.
‘Late onset’-hypogammaglobulinemie
‘Common variable immunodeficiency’ (CVID) of ‘late
onset’-hypogammaglobulinemie is een immunologisch zeer
heterogene groep van soms familiair, soms sporadisch
voorkomende aandoeningen waarbij geleidelijk aan een
progressieve immuundeficiëntie ontstaat.
CVID wordt gediagnostiseerd bij kinderen van 18 maanden tot 5
jaar, met een ‘tweede piek’ bij
kinderen rond de puberteit. Vaak leidt de ziekte echter pas in
de tweede of derde levensdecade tot klinische problemen (14).
Secundair-lymfoïde organen zijn wel aanwezig. Vaak is er zelfs
een lymfadenopathie door reactieve folliculaire hyperplasie.
Het is uiterst moeilijk om de diagnose van CVID definitief te
stellen, en het is belangrijk om beter gedefinieerde
immuundeficiënties zoals agammaglobulinemie, hyper-
IgM-syndroom of X-gebonden proliferatief syndroom uit te
sluiten.
CVID lijkt voor te komen met een incidentie van 1:50.000 en is
gelijkelijk verdeeld over vrouwen en mannen. Het ziektebeeld
wordt gekenmerkt door hypogammaglobulinemie waarbij met
name de serumconcentraties van IgG (< 4 g/l) en veelal IgA
verlaagd zijn tot minder dan de gemiddelde
waarde minus tweemaal de standaarddeviatie van gezonde
personen met dezelfde leeftijd. IgM kan
in normale maar ook sterk verlaagde concentraties aanwezig
zijn. CVID wordt gekarakteriseerd door
een afwezigheid van een significante antistofrespons na
vaccinatie met zowel eiwit als polysaccharideantigenen.
Het aantal B-cellen in de circulatie kan variëren van verlaagd
tot normaal. Het aantal T-cellen is
meestal normaal, met bij sommige patiënten een verlaagd aantal
CD4+-T-cellen (met name de CD45RA+-naïeve cellen) en bij
anderen een verhoogd aantalCD8+-T-cellen.
Als oorzaak voor CVID zijn functionele defecten op het niveau
van de B-cellen, T-cellen of antigeenpresenterende cellen
gesuggereerd.
Moleculaire defecten die ten grondslag liggen aan CVID waren
echter tot voor kort niet beschreven. Recent is
ICOS-deficiëntie als oorzaak voor CVID aangetoond bij 4
patiënten uit 2 families (15). ICOS is een co-stimulatoir
molecuul betrokken bij de interactie tussen geactiveerde T- en
B-lymfocyten in de secundaire lymfoïde organen.
Afwezigheid van ICOS lijkt geen invloed te hebben op de
intrinsieke functie van T-lymfocyten. Daarentegen lijken het
aantal naïeve B-cellen, het aantal B-cellen met een
immunoglobuline-
klasseswitch en het aantal geheugen-B-cellen verlaagd te zijn
bij patiënten met een CVID-achtig
ziektebeeld en ICOS-deficiëntie.
Bij patiënten met CVID lijkt er sprake te zijn van
immuundisregulatie, waarbij auto-immuunziekten en
maligniteiten (zoals lymfomen) in verhoogde frequentie
voorkomen. Zoals reeds genoemd heeft CVID een progressief
beloop. In de loop van de tijd ontwikkelen zich progressief
IgA-deficiëntie en IgGsubklassedeficiëntie.
Antipolysaccharide-antistofdeficiëntie lijkt al vroeg op te
treden. Later kan dit uitbreiden tot een hypogammaglobulinemie
met gestoorde antistofresponsen op eiwitantigenen, en
eventueel een
T-lymfopenie met gestoorde T-cel-gemedieerde immuniteit.
De infectieuze problemen blijken vaak eerder aanwezig te zijn
dan de laboratoriumafwijkingen.
Vandaar dat herhaald onderzoek bij blijvende klinische
problemen ondanks eerdere normale bevindingen
essentieel is om de diagnose tijdig te stellen. Vooral als in
de familie meerdere personen met al dan niet
milde immuundeficiëntie voorkomen (zoals IgA-deficiëntie), is
het verstandig actief naar deze diagnose te
blijven zoeken bij een kind dat infectieuze problemen
houdt.
Patiënten met CVID presenteren zich vaak met infecties van de
bovenste en onderste luchtwegen door
gekapselde extracellulaire bacteriën en met gastrointestinale
problemen die veroorzaakt kunnen worden
door infecties (Giardia, Salmonella, Shigella, Cam
pylobacter). Darmproblemen (chronische diarree,
malabsorptie en eiwitverlies via de darm) worden echter ook
regelmatig gezien zonder dat er sprake is
van een darminfectie.
Dit wordt geïnterpreteerd als een teken van T-cel-disregulatie
en auto-immuniteit tegen het darmepitheel. Opportunistische
infecties zijn bij CVID zeldzaam. Bij 20% van de patiënten met
CVID worden tekenen van auto-immuunziekten gezien zoals
auto-immuun hemolytische anemie en idiopathische trombopenie.
Een mogelijk andere subgroep van CVID (20-30%) onderscheidt
zich door de aanwezigheid van granulomen in de lever, longen,
lymfeklieren, of de huid, vaak in combinatie met splenomegalie
en lymfadenopatie.
Patiënten met CVID worden behandeld middels intraveneuze of
subcutane suppletie van de immunoglobulinen.
Dit leidt tot een verminderd optreden van infecties, maar
heeft geen effect op de niet-infectieuze
darmklachten en de kans op het ontwikkelen van maligniteiten
waaraan mogelijk de T-cel-disfunctie ten
grondslag ligt.
Ned Tijdschr Klin Chem Labgeneesk 2004; 29: 145-150
Complement en ontsteking
A.J. HANNEMA1 en C.E. HACK2
Het complementsysteem omvat meer dan 30 plasma en
membraaneiwitten die een taak hebben bij de verdediging van
het lichaam tegen pathogene microorganismen.
Naast hun rol bij de opsonisatie van deze organismen, leidt
complementactivatie tot het ontstaan van producten met tal van
pro-inflammatoire effecten zoals bijvoorbeeld chemotaxie.
Tot voor kort werd het complementsysteem onderscheiden in twee
activatieroutes, de klassieke en de alternatieve
activatieroute. Recent is daar een derde activatieroute, de
lectine-activatieroute, aan toegevoegd.
In dit artikel wordt aan de hand van de drie verschillende
activatieroutes de rol van complement als ontstekingsmediator
besproken. Hierbij wordt ook ingegaan op afwijkende
complementactivatie in relatie
tot ziekte en op potentieel therapeutische mogelijkheden van
complementfactoren.
Ontstekingen hebben tot doel om binnendringende
micro-organismen of beschadigd weefsel op te ruimen.
Ontstekingen worden gekenmerkt door pijn, lokale roodheid en
verhoging van de temperatuur als
gevolg van vasodilatatie, en door zwelling als gevolg van
oedeemvorming en migratie van leukocyten naar
de plaats van ontsteking.
Systemisch kunnen er ook verschijnselen optreden, zoals
veranderingen van het eiwitspectrum, een verhoging van de
lichaamstemperatuur en hormonale veranderingen. Deze
systemische verschijnselen zijn bekend als de
acutefasereactie.
Ontstekingsreacties ontstaan als gevolg van het vrijkomen en
activeren van ontstekingsmediatoren. Cytokinen worden
als de belangrijkste ontstekingsmediatoren beschouwd, omdat
zij de gebeurtenissen die
plaats vinden wanneer micro-organismen het lichaam
binnendringen, of wanneer weefsel beschadigd wordt,
voor een belangrijk deel regisseren. Naast cytokinen zijn er
nog vele andere ontstekingsmediatoren, waaronder het
complementsysteem.
Ontstekingsreacties zijn niet altijd onschuldig, maar kunnen
zelf ook schade aan de weefsels berokkenen.
Bij tal van humane ziekten, zoals sepsis, artritis,
vasculitis, CARA, inflammatoire darmziekten, lijkt dit
negatieve effect van ontstekingsreacties te domineren.
Het is daarom van belang de precieze rol van de verschillende
ontstekingsmediatoren bij dergelijke ziekten
te kennen om gericht ingrijpen in het ontstekingsproces
mogelijk te maken. Het doel van dit artikel is inzicht te
verschaffen in de rol van complement als ontstekingsmediator
en het nut van complementbepalingen
in de diagnostiek bij infectie en ontstekingsreacties.
Tevens wordt besproken hoe manipulatie van het
complementsysteem therapeutisch nuttig kan zijn.
Complement als ontstekingsmediator
Het complementsysteem is een verzameling van ruim 30 plasma-
en membraaneiwitten die een taak hebben bij de verdediging van
het lichaam tegen pathogene micro-organismen. Een belangrijke
functie van complement tijdens ontstekingsreacties is het
merken (opsonisatie) van deze organismen voor fagocyterende
cellen, zoals macrofagen en granulocyten.
Laatstgenoemde cellen bezitten verschillende receptoren welke
fragmenten van complementfactoren herkennen die op bacteriën
zijn gebonden. Via deze receptoren worden door complement
gemerkte organismen gebonden en gefagocyteerd.
De belangrijkste complementfactor verantwoordelijk voor
opsonisatie is C3, oftewel de derde complementfactor.
Complementactivatieproducten zoals C3a en C5a, en
C5-9-complexen bevorderen chemotaxie, leiden tot activatie en
degranulatie van neutrofielen en mestcellen, verhoging van
endotheelpermeabiliteit, inductie van adhesiemoleculen zoals
ICAM-1 op endotheel, contractie van gladde spiercellen in de
bronchiaalboom, relaxatie van gladde spiercellen in arteriolen
en versterking van cytokinenproductie door macrofagen.
Insertie van C5-9-complexen in een celmembraan kan tot lysis
van de targetcel of bacterie leiden.
Activatieroutes
Slechts één decennium geleden werd nog algemeen geaccepteerd
dat er twee activatieroutes van complement bestonden: de
klassieke route, die geactiveerd werd door Fc-fragmenten van
de immunoglobulines IgG en IgM in antistof-antigeencomplexen
en de alternatieve route, waarvan de activatie direct via
membraanstructuren op de bacteriecelwand plaatsvindt.
De eerste route behoort tot de verworven immuniteit, omdat
hierbij eerst een antistofrespons tegen een pathogeen opgewekt
moet worden, de tweede route maakt onderdeel uit van de
aangeboren immuniteit,
die onmiddellijk kan reageren op lichaamsvreemde structuren,
in de vorm van suikers, die op de bacteriecelwand herkend
worden.
De klassieke route kan ook door eiwitten als C-reactief
proteïne (CRP) geactiveerd worden, en vormt dan weer een
effectormechanisme van het aangeboren immuunsysteem.
De derde activatieroute, eveneens onderdeel uitmakend van de
aangeboren immuniteit, is de lectineroute.
Deze route wordt geactiveerd door binding van het eiwit MBL
(mannosebindend lectine) aan bacteriën
of andere micro-organismen met mannose op het oppervlak. MBL
lijkt qua structuur op C1q, het eiwit van de klassieke route
dat aan immuuncomplexen bindt, en heeft een collageenachtig
deel en een lectine deel , dat in staat is aan suikers van de
bacteriecelwand te binden. Er zijn inmiddels meer MBL-achtige
eiwitten
bij de mens ontdekt, zoals ficoline.
Activatie van de lectineroute kan dus vermoedelijk door
verschillende eiwitten geïnduceerd worden.
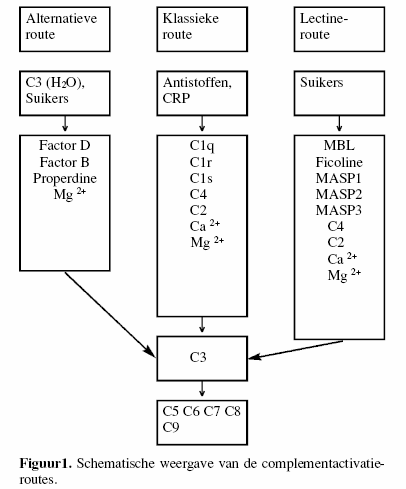
De verschillende eiwitten betrokken bij de activatie van de
verschillende complementroutes staan aangegeven in figuur 1.
Alternatieve route
Deze wijze van activatie werd fylogenetisch als de oudste
complementroute gezien, maar tegenwoordig
denkt men dat de lectineroute feitelijk eerder ontstond
tijdens de evolutie. Alternatieve-route-activatie
leidt tot fixatie van een afbraakproduct van C3, C3b, op
pathogene organismen. In natief C3 zit een interne
thio-ester die na activatie van het molecuul tot C3b aan de
oppervlakte komt en via hydroxyl- of aminogroepen covalent aan
een activator kan fixeren. Het merendeel van de C3b-moleculen
zal echter niet
fixeren aan de activator omdat watermoleculen ook efficiënt
met de thio-ester reageren en deze hydrolyseren voordat C3b
gefixeerd is, wat leidt tot ‘fluidphase’ C3b. Zelfs in natieve
C3-moleculen blijkt de
thio-ester na verloop van tijd door water te worden
gehydrolyseerd. Dit gehydrolyseerde C3 lijkt qua conformatie
erg op C3b en kan met factor B en D reageren waardoor een
C3Bb-complex ontstaat dat weer
natief C3 omzet in C3a en C3b.
Via dit mechanisme wordt er dus continu een klein beetje C3
geactiveerd (de zogenaamde ‘tick-over’ van C3). De factoren H
en I voorkomen dat C3 te erg wordt geactiveerd: factor H door
Bb uit het C3bBb-complex te verdringen, en I door in het
C3bH-complex C3b te inactiveren tot C3bi.
Een alternatieve-route-activator is een stof die een beetje
C3b op zodanige wijze bindt dat dit beschermd
is tegen de remmers I en H, en een interactie met B en D kan
aangaan.
Deze interactie leidt tot de vorming van een C3bBb-complex op
de activator wat weer meer natief C3 activeert, dat weer
fixeert, factor B bindt, enz.
Aangezien, zoals gezegd, deze activatie niet te controleren is
door de remmers I en H, leidt fixatie van een klein beetje C3b
op zo’n activator ineens tot een sterke C3-activatie.
Polysacchariden op bacteriën, gistcellen, schimmels e.d.
kunnen op een dergelijke wijze uitstekend de alternatieve
route activeren.
C3b dat op lichaamscellen van de mens fixeert, wordt
geïnactiveerd door factor H, waardoor verdere activatie stopt.
Hierdoor kan geen schade aan de eigen cellen aangericht
worden, ook al bevinden ze zich dicht
bij de bron van de activatie. Verdergaande activatie van C3
leidt uiteindelijk tot de fixatie van een extra
C3b-molecuul op het C3bBb-convertase, dat daardoor van
specificiteit verandert: het gaat nu C5 in
plaats van C3 activeren.
C5-activatie leidt uiteindelijk tot de vorming van het zgn.
MAC (‘membrane attack complex’) dat is samengesteld uit de
complementfactoren C5b, C6, C7, C8 en meestal meerdere
moleculen
C9.
Het MAC nestelt zich in de lipidenlaag van de
bacteriemembraan, die daardoor permeabel wordt.
Door het grote verschil in osmotische druk binnen en buiten de
cel zal deze vloeistof aanzuigen en barsten
(bacteriolysis) en wordt de bacterie vernietigd.
Naast de remmers I en H, en de factoren B, D en C3b, kent de
alternatieve route nog een factor, P (properdine), die zorgt
voor stabilisatie van het C3bBbcomplex, waardoor dit langer
werkzaam kan zijn. P is
dus in dit opzicht een tegenpool van factor H.
Bij de verschillende activatiestappen waarbij
complementfactoren enzymatisch gesplitst worden, ontstaan
kleine fragmenten die een sterke biologische werking hebben,
zoals de anafylatoxinen C3a en C5a. Dit zijn
chemotactisch actieve stoffen die leukocyten naar de plaats
van de ontsteking dirigeren. Het C3b zelf is, behalve een
onderdeel van het alternatieve-route-convertase, ook een
opsonine dat verantwoordelijk is voor
binding aan complementreceptoren op fagocyterende cellen. Het
legt hiermee een verbinding met de cellulaire immuniteit via
het fagocytoseproces.
In het laboratorium wordt de activiteit van deze route gemeten
d.m.v. de AP50-titerbepaling, waarbij konijnen - of
cavia-erytrocyten in afwezigheid van Ca2+ door complement
gelyseerd worden. De afwezigheid van
C3, factor H, factor I of factor D zal leiden tot een sterk
verlaagde titer. Het ontbreken van factor P leidt
eveneens tot een verlaagde AP50-titer, echter minder sterk dan
bij factor-H-, -I-, of -D-deficiëntie. Verder
onderzoek van de afzonderlijke alternatieve-route-factoren kan
van belang zijn om deficiënties hiervan
vast te stellen.
Het is bekend dat het ontbreken van een van de
alternatieve-route-factoren kan leiden tot
meningococcen-ziekte. Ook in Nederland zijn deze deficiënties,
gepaard gaande met infectie met Neisseria meningitidis bekend:
factor H (1), factor D (2), factor P (3, 4). Ook twee
factor-I-deficiënties zijn inmiddels
bekend geworden.
Deze deficiëntie gaat eveneens gepaard met recidiverende
meningococceninfecties.
Alvorens onderzoek naar een factor-H- of -I-deficiëntie te
doen is het gewenst de concentraties
van C3, factor B en het afbraakproduct C3d te meten.
Bij de factor-H-deficiëntie zullen C3 en factor B (sterk)
verlaagd zijn, terwijl de C3d-concentratie juist
sterk verhoogd is ten gevolge van een continue,
ongecontroleerde afbraak van C3, vanwege het ontbreken
van factor H (de ‘tick-over’ van C3, zie boven, is
ongeremd).
Bij een factor-I-deficiëntie zien we iets dergelijks voor C3
en factor B, maar in dit geval is er geen toename van de
C3d-concentratie. Dat vindt zijn oorzaak in het feit dat
factor I noodzakelijk is voor de afbraak van C3b in C3c en
C3d.
Zonder aanwezigheid van factor I vindt deze splitsing niet
plaats (5).
Behalve de associatie met meningococceninfecties is de
deficiëntie van factor H betrokken bij een atypische vorm van
het hemolytisch-uremisch-syndroom (HUS). HUS wordt
gekarakteriseerd door hemolytische anemie, trombocytopenie,
diarree en acute nierinsufficiëntie.
Bij de atypische vorm, waarbij vooral puntmutaties gevonden
worden in het C-terminale
deel van de factor-H-keten (78%) of door mutatie een stopcodon
ontstaat (22%), is er geen voorafgaande
diarree.
Deze mutaties, die in veel gevallen een erfelijk karakter
hebben, leiden tot een inactieve vorm van factor H, of ze
blokkeren de uitscheiding van factor H, waardoor het zich in
de cel ophoopt.
Omdat de gebruikelijke testmethoden geen onderscheid kunnen
maken tussen de actieve en de inactieve vorm, zal een
afwijkende (verlaagde) concentratie lang niet in alle gevallen
gevonden worden (6).
In tegenstelling tot de meeste andere deficiënties van
complementfactoren zijn de patiënten met atypische HUS meestal
heterozygoot en maken ze naast de afwijkende vorm ook normaal,
functioneel-actief factor H aan.
Klassieke route
Later ontstaan in de evolutie, maar veel bekender door de
vroege ontdekking door Bordet in 1895, is de
klassieke complementactivatieroute. Deze is afhankelijk van
het opwekken van specifieke antistoffen tegen
organismen die het lichaam binnengedrongen zijn.
Hierdoor komt dit verdedigingsmechanisme later (± 5-7 dagen)
op gang dan de alternatieve route. Het
werkt echter veel efficiënter, omdat alleen immuuncomplexen en
cellen waaraan IgM, IgG1, IgG2 of
IgG3 gebonden is, door complement herkend en opgeruimd
worden.
De reactievolgorde, te zien in figuur 1, loopt via de binding
van C1q aan het Fc-fragment van immunoglobulines en activatie
van het aan C1q gebonden C1r en C1s. Geactiveerd C1s splitst
C4 en C2, waarbij het C3-convertase C4b2a gevormd wordt.
C1-esteraseremmer zorgt ervoor dat na de activatie door
immuuncomplexen de activatie beperkt blijft tot de directe
omgeving van het complex en zich niet over het gehele
bloedvolume uitbreidt.
Tekort aan C1-esteraseremmer, zoals dat bij het hereditair
angio-oedeem gevonden wordt, leidt tot het vrijkomen van grote
hoeveelheden vasoactieve peptiden, deels uit geactiveerde
complementfactoren, maar deels ook uit het
contactactivatiesysteem van de intrinsieke stollingsroute.
Dit laatste wordt verklaard uit het feit dat C1-
esteraseremmer ook de belangrijkste remmer is van
kallikreïne en geactiveerde Hageman-factor (factor XII), de
twee actieve proteasen van het contactacitvatiesysteem van de
bloedstolling.
De excessief gevormde vasoactieve peptiden geven
vaatverwijding en oedeemvorming.
Het klassieke-route-convertase C4b2a splitst C3, waarbij het
grootste fragment C3b voor een deel via de
thio-ester aan het oppervlak van het micro-organisme of
immuuncomplex gebonden blijft en via complementreceptoren op
b.v. granulocyten tot fagocytose leidt. Net als bij de
alternatieve route zal bij verdergaande activatie van C3 op
een gegeven moment een C3b-molecuul op het C4b2a-convertase
fixeren, waardoor de specificiteit van laatstgenoemde
verandert en het C5 gaat omzetten. Dit leidt uiteindelijk ook
tot de vorming van het MAC.
Voorts is op te merken dat het C3b dat op een klassieke
activator fixeert, ook een interactie met factoren B en D kan
aangaan waarbij net als boven beschreven voor de alternatieve
route via het C3bBb-convertase natief C3 geactiveerd kan
worden. De alternatieve route amplificeert dus als het ware op
C3-niveau de activatie van de klassieke route.
Omdat bacteriën en immuuncomplexen complement kunnen
activeren, gaat men er meestal van uit dat zij
de oorzaak van complementactivatie bij ontstekingsziekten
zijn. Toch wordt er bij een aantal ziektebeelden
complementactivatie gevonden, zonder dat deze activatoren
aanwezig zijn.
Activatie van complement tijdens behandeling van
kankerpatiënten met interleukine-2 is een voorbeeld hiervan.
Deze patiënten ontwikkelen (als ongewenst bijeffect) een
gegeneraliseerde ontstekingsreactie op de toediening van dit
interleukine, welke het gevolg is van het vrijkomen van TNF
(7) en IL-6. Opmerkelijk genoeg wordt bij deze patiënten ook
een sterke complementactivatie gevonden. Er is hier echter
geen sprake van een infectie door bacteriën, of aanwezigheid
van immuuncomplexen.
Aangetoond kon worden dat de complementactivatie in dit geval
door het eiwit C-reactief proteïne (CRP) gemedieerd wordt.
CRP is een acuutfase-eiwit dat o.a. aan fosfocholine kan
binden. Fosfocholine komt voor in koolhydraten
van bacteriekapsels en op lipopolysachariden van
micro-organismen, zoals S. pneumoniae, Haemophilus
influenzae, Neiserria meningitides en Aspergillus fumigatus.
Gebonden CRP kan vervolgens, net als een
IgG- of IgM-antistof, de klassieke complementroute activeren.
Ook wordt fosfocholine gevonden in fosfatidylcholine, een
fosfolipide van celmembranen.
In de membraan van gezonde cellen is het fosfocholine niet
bereikbaar voor CRP, maar op necrotische cellen en ook op
reversibel beschadigde cellen kan CRP wel aan het fosfocholine
in de membraan binden.
Deze bereikbaarheid van fosfocholine heeft te maken met de
fosfolipidensamenstelling van de celmembraan.
Het aan een ligand gecomplexeerd CRP wordt herkend door C1q en
via de vorming van C3-convertase
activeert dit de klassieke route. Het lijkt erop dat er voor
C1q-binding meerdere CRP-moleculen in elkaars
directe omgeving nodig zijn, zoals dat ook geldt voor de
activatie van complement door IgGmoleculen.
Het vermogen van CRP zich te binden aan pathogene organismen
en het verwijderen hiervan
door complementactivatie en rekrutering van fagocyterende
cellen is belangrijk voor de aangeboren immuniteit.
Verder heeft het een duidelijke rol in het opruimen van
necrotische en apoptotische cellen en draagt zo bij aan het
herstel van beschadigde weefsels (8).
Activatie van de klassieke complementroute door beschadigde en
apoptotische cellen, al dan niet door adaptormoleculen als
CRP, kan op verschillende manieren tot ziekten leiden.
Excessieve activatie
speelt een rol bij bijvoorbeeld het acute myocardinfarct, te
geringe activatie leidt tot het ziektebeeld systemische lupus
erythematosus (SLE).
Beide beelden worden hieronder besproken.
Bij het acuut myocardinfarct (AMI) raken cardiomyocyten
ischemisch waardoor de fosfolipidensamenstelling van hun
celmembraan verandert. De normale asymmetrie van hun
lipidenmembraan, die ontstaat omdat de fosfolipiden aan de
binnen- en buitenzijde van de membraan onder normale
omstandigheden niet hetzelfde zijn, kan niet worden
gehandhaafd. Het gevolg hiervan is dat deze cellen negatief
geladen fosfolipiden in hun buitenmembraan krijgen. Secretoir
fosfolipase-A2 (sPLA2) kan hierdoor de membranen van
beschadigde en dode cellen hydrolyseren, waarbij CRP zich aan
de gevormde lysofosfolipiden bindt en het complement systeem
activeert (figuur 2).
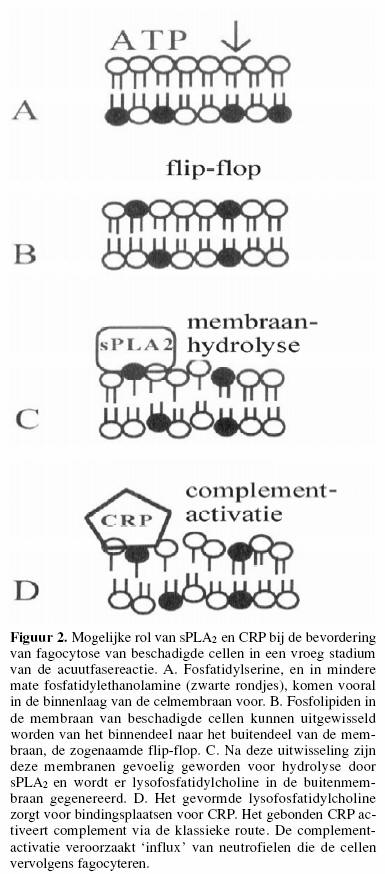
Oxidatie van de fosfolipiden kan een vergelijkbaar effect
hebben. Fagocyten zullen zo worden aangezet
de met complement gemerkte cellen op te ruimen met als gevolg
een verergering van de ontsteking (9).
Lagrand et al. (10) onderzochten 17 patiënten die overleden
waren aan AMI. Zij vonden depositie van
CRP, tezamen met de complementfactoren C3 en C4, in het
geïnfarcteerde gebied en niet in normaal uitziend weefsel van
het myocardium (figuur 3). Nijmeijer et al. (11) vonden bij 56
patiënten die aan een
acuut myocardinfarct waren overleden een duidelijke correlatie
tussen geactiveerd complement in het aangedane weefsel en de
aanwezigheid van CRP-complement complexen welke specifiek
worden gevormd
tijdens activatie van complement door CRP.
Deze gegevens duiden erop dat CRP een belangrijke rol speelt
bij activatie van complement in het myocardweefsel tijdens een
hartinfarct.
Dit onderzoek is van belang bij het zoeken naar therapeutische
mogelijkheden om de weefselbeschadiging bij AMI te beperken.
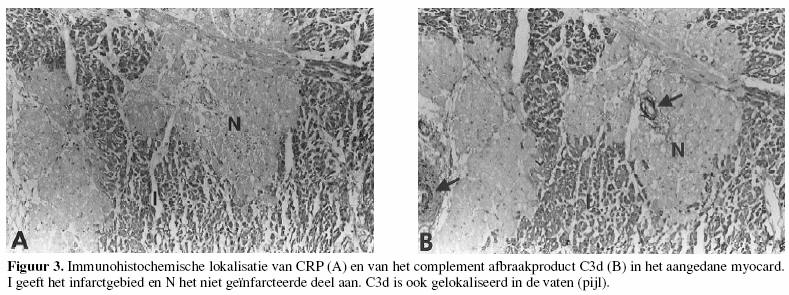
Zo was uit dierexperimenteel onderzoek bij honden gebleken dat
de toediening van hoge dosis C1-esteraseremmer de uitbreiding
van het necrotische gebied van het myocard doet verminderen.
De Zwaan
et al. (12) toonden aan dat bij 22 patiënten met een
hartinfart de vorming van de C4-afbraakproducten
C4b/c na toediening van C1-esteraseremmer significant afnam in
vergelijking met een controlegroep.
Nog belangrijker was de bevinding dat patiënten die
C1-esteraseremmer toegediend hadden gekregen een
kleiner infarct hadden dan historische controles. Dat duidt
erop dat remming van complementactivatie bij
patiënten met AMI therapeutisch zinvol kan zijn. Dit gunstige
effect wordt gevonden zelfs als de remmer
pas 6 uur na optreden van het AMI wordt toegediend, wat
suggereert dat remming van complementactivatie
zelfs in een relatief laat stadium nog zinvol kan zijn.
Niet alleen teveel, maar ook te geringe activatie van
complement door apoptotische cellen kan tot ziekte
leiden. Reeds lang is bekend dat genetische deficiënties van
de klassieke route van het complementsysteem een sterke
associatie vertonen met het ontwikkelen van SLE. Feitelijk
hebben personen met een deficiëntie van het C1-complex (C1q,
C1r of C1s) vrijwel 100% kans om SLE te ontwikkelen.
Men heeft altijd gedacht dat de ontwikkeling van SLE bij een
tekort aan een van de C1-onderdelen komt door een gestoorde
opruiming van immuuncomplexen.
Sinds een paar jaar is bekend dat apoptotische cellen ook de
klassieke route van het complementsysteem
activeren. Bovendien is gebleken dat vele auto-antistoffen die
bij SLE voorkomen feitelijk antistoffen zijn tegen afval van
apoptotische cellen (o.a. nucleosomen en fosfolipiden).
Derhalve denkt men nu dat deficiënties van de klassieke route
leiden tot een verminderde opruiming van apoptotische cellen,
waardoor
deze het specifieke immuunsysteem stimuleren tot de vorming
van antistoffen. Die antistoffen vormen
vervolgens immuuncomplexen, die met betrokken auto-antigenen
immuuncomplexen vormen die in de vaten neerslaan en daar via
fagocyterende cellen en mogelijk ook de alternatieve route van
het complementsysteem tot ontstekingen leiden.
In een dergelijk scenario is te verwachten dat ook
deficiënties van CRP en serumamyloïdcomponent P (SAP), die
immers als adaptormoleculen functioneren voor de activatie van
complement door apoptotische cellen, tot SLE leiden. Recente
studies in muizen duiden daar inderdaad op.
Lectineroute
Tamelijk recent in de geschiedenis van het complementonderzoek
werd ontdekt dat het mannosebindend
lectine (MBL) een component was van een nieuwe
complementactivatieroute. Hierbij wordt zonder
medewerking van antistof, maar ook met uitsluiting van C1, de
klassieke route in gang gezet, te beginnen met C4 en C2.
Het in de lever gesynthetiseerde MBL heeft een oligomere
structuur en bestaat uit drie identieke peptideketens
die elk een Ca2+ -afhankelijk en polysacharidebindend
lectinedeel, een hydrofoob deel, een collageendeel en een
cysteïnerijk N-terminaal deel bezitten.
Tezamen vormen de drie ketens een klassieke
tripelhelixstructuur. Het MBL kan in verschillende oligomere
structuren voorkomen. Tetrameren zijn nodig voor activatie van
complement, di- of trimeren hebben vermoedelijk geen
biologische activiteit.
MBL bindt aan verschillende suikers zoals mannose,
N-acetylgalactosamine, N-acetylglucosamine en maltose.
Door de lage bindingsaffiniteit van de afzonderlijke
bindingsplaatsen is het noodzakelijk dat meerdere lectinedelen
van één MBL-molecuul tegelijkertijd aan de repeterende
suikerstructuren op micro-organismen hechten om een sterke
functionele binding te krijgen. Wat functie betreft lijkt MBL
sterk op C1q. Beide behoren dan ook tot een familie van
structureel verwante eiwitten.
Recent is bekend geworden dat ook verschillende andere leden
van deze familie, zoals de verschillende ficolines, het
complementsysteem kunnen activeren.
De gemiddelde concentratie van MBL in plasma is ±1,7 µg/ml,
maar kan sterk variëren. De concentratie is erfelijk bepaald
en wordt sterk beïnvloed door polymorfismes in exon 1 van het
MBL-gen op chromosoom 10.
De polymorfismen worden aangetroffen in codon 52
(argininecysteïne), codon 54 (glycine→asparaginezuur) en codon 57 (glycine→glutaminezuur ) .
Er is sprake van een opmerkelijke geografische variabiliteit
bij de polymorfismen; de codon-52- variant wordt hoofdzakelijk
in het Eurazische gebied gevonden en de codon-57-variant
vooral in Afrikaanse populaties ten zuiden van de Sahara. Ook
z.g. promotorpolymorfismen van het MBL-gen, speciaal een
basenpaarsubstitutie in codon –221 (GC), kunnen
leiden tot zeer uiteenlopende en vaak verlaagde concentraties.
Het gevolg van deze polymorfismen is dat de secundaire
structuur van de tripelhelix niet gevormd kan worden en er een
niet functionerend eiwit ontstaat. Hierdoor is zelfs bij
heterozygoten de plasmaconcentratie van het functioneel
actieve (suikerbindende) molecuul slechts 1/8 van de normale
waarde. Deficiënties zijn dan ook bepaald niet zeldzaam: bij
±15-20% van de bevolking wordt een sterk verlaagde
concentratie gevonden.
Voor het meten van de MBL-concentratie is het van belang een
functionele bepaling te doen die gebruik maakt van binding aan
mannose, daar anders de niet functionerende MBL-moleculen mee
bepaald worden.
Omdat MBL als opsonine van diverse bacteriën functioneert en
tevens complementactiverende eigenschappen
heeft, levert een MBL-deficiëntie een verhoogd risico op
infectieziekten op. Een MBL-deficiëntie in geïsoleerde vorm
hoeft geen belangrijk gezondheidsrisico op te leveren, maar
kan problemen opleveren bij individuen waarbij andere
deficiënties van het immuunsysteem aangetroffen worden, zoals
mucoviscoïdosis of een neutropenie als gevolg van het gebruik
van cytostatica.
Vanwege de hoge frequentie van MBL-deficiënties, is het
moeilijk te begrijpen dat deze mutaties in de loop van de
evolutie niet verdwenen zijn, omdat de betrokken
individuen in het nadeel zijn t.o.v. soortgenoten. Het
is echter bekend dat bepaalde micro-organismen die zich binnen
de cel vermenigvuldigen mogelijk gebruik maken
van complementactivatie door MBL om de gastheercellen
(fagocyten) binnen te dringen. Dit is onder
meer het geval bij Mycobacterium tuberculosis, M. leprae en
Leishmania-soorten.
In gebieden waar de ziekten tengevolge van deze pathogene
organismen voorkomen, zullen individuen met een lage
MBLconcentratie in het voordeel zijn. Het is dus aantrekkelijk
om te veronderstellen dat MBL-deficiëntie
onder bepaalde omstandigheden ook gúnstig kan zijn, namelijk
bescherming biedt tegen intracellulaire
ziekteverwekkers (13, 14).
De activatie van complement via MBL wordt in gang gezet door
de meervoudige binding van MBL aan
repeterende suikers van oppervlaktestructuren van bacteriën,
schimmels en virussen (15). Door associatie
van MBL met de serineproteases MASP-1, MASP-2 en MASP-3, die
wat structuur betreft op de
complementfactoren C1r en C1s lijken, worden C4 en C2
geactiveerd, waardoor het klassieke-route-C3-
convertase-C4b2a gevormd wordt. Met name MASP-2 lijkt sterk op
C1s in zijn C4- en C2-activerende eigenschappen, waarbij
opgemerkt moet worden dat dit serineprotease niet zoals C1s
via een andere factor
(C1r) geactiveerd wordt, maar direct na binding van MBL aan
een substraat zijn enzymatische activiteit
t.o.v. C4 en C2 verkrijgt (16). De C3-activatie vindt
vervolgens op de gebruikelijke wijze plaats.
Conclusie
Het complementsysteem is een belangrijke mediator van de
humorale immuniteit en ontstekingsreacties,
waarbij het via opsonisatie een brugfunctie vervult tussen
humorale en cellulaire afweer. De alternatieve en
lectineroute zijn direct te activeren via suikerstructuren op
pathogenen, terwijl de klassieke route pas na het
opwekken van antistoffen of CRP op gang komt, maar hiermee wel
een grotere selectiviteit en hogere efficiëntie
bezit. Hoewel een goed functionerend complementsysteem als
onderdeel van de afweer tegen ziekteverwekkers
voordelig is voor het individu, kan het in andere gevallen
doorslaan in zijn werking en daarmee
verergering van ontstekingsreacties geven of het binnendringen
van micro-organismen bevorderen.
Stoornissen betreffende het immuunsysteem en het
verdedigingssysteem

|
Hoofdmenu |
