|
Ned Tijdschr Klin Chem 2000; 25: 13-27
Diagnostiek van lysosomale stapelingsziekten
J.G.N. de Jong, R.A Wevers, C.J.M. van den Berg, M.L.F.
Liebrand-van Sambeek, A.A.E.T. van Rens en H.G.M. Roelofs
Aan de meeste lysosomale stapelingsziekten ligt een deficiëntie
van één van de lysosomale enzymen betrokken bij de afbraak van
macromoleculen tot de monomoleculaire componenten ten
grondslag.
De drie belangrijkste groepen, ingedeeld op basis van het
belangrijkste stapelingsproduct, zijn de sfingolipidosen,
oligosaccharidosen en mucopolysaccharidosen.
Mucolipidose II en III worden veroorzaakt door een defect in de
processing van een aantal lysosomale enzymen, waardoor meer
enzymen tegelijkertijd deficiënt zijn.
Ook defecten in het transport over de lysosomale membraan, zoals
van neuraminezuur, worden tot de lysosomopathieën gerekend.
Van enkele neuronale ceroid lipofuscinosen is nu aangetoond, dat
het lysosomale stapelingsziekten zijn.
De kliniek van de lysosomopathieën is heterogeen, met als
belangrijkste kenmerken, knik in de psychische en/of motore
ontwikkeling en hepato- en/of splenomegalie.
Screening in urine is mogelijk voor een tiental oligosaccharidosen
door analyse van oligosacchariden en voor de groep van de
mucopolysaccharidosen door meting van het
glycosaminoglycaangehalte in de urine.
Een defect in het transport van neuraminezuur resulteert in een
verhoogde uitscheiding van deze suiker in de urine.
Bij een afwijkend patroon van oligosacchariden of een verhoogde
uitscheiding van glycosaminoglycanen worden de betreffende enzymen
in leukocyten, geïsoleerd uit een bloedmonster, gemeten om het
defect te bevestigen of uit te sluiten.
Voor het grootste deel van de sfingolipidosen en voor de neuronale
ceroid lipofuscinosen is geen voorscreening mogelijk.
De sfingolipidosen en nu ook enkele neuronale ceroid
lipofuscinoses kunnen worden bevestigd of uitgesloten door directe
meting van de diverse enzymen in leukocyten en/of fibroblasten.
Het lysosoom als celorganel is voor het eerst beschreven door de
De Duve. Het is betrokken bij de afbraak van diverse cellulaire
macromoleculen zoals lipiden, glycoproteïnen en
glycosaminoglycanen tot de monomoleculaire componenten.
De afbraak wordt verzorgd door katabole enzymen, zelf ook
glycoproteïnen, met pH optima in het zure gebied.
Is de afbraak gestoord door een deficiëntie van één van de
lysosomale enzymen dan ontstaat door een blokkade in het
afbraakproces een opeenhoping van cellulair materiaal. Dit
materiaal hoopt zich op in de lysosomen die uitdijen tot vacuolen
en vaak al microscopisch waarneembaar zijn.
Klinisch brengt dit met zich mee dat orgaanvergrotingen als
hepato- en splenomegalie gevonden kunnen worden.
Het enzymdefect kan ook in het centrale zenuwstelsel tot uiting
komen. Verregaande cerebrale degeneratie is vaak het gevolg. Niet
zelden gaat een dergelijk lysosomaal enzymdefect gepaard met een
duidelijke knik in de geestelijke- en/of motorische ontwikkeling.
Ook het fenotype van de patiënt kan aanleiding zijn om aan een
lysosomale stapelingsziekte te denken. Het is onjuist aan te nemen
dat het uitsluitend gaat om een groep van ziekten die zich slechts
op jeugdige leeftijd kan manifesteren. Voor de meeste lysosomale
afwijkingen is dit wel het geval, maar soms komen duidelijk adulte
vormen voor.
Bekende voorbeelden van lysosomale ziekten die in een infantiele
maar ook in een adulte vorm kunnen voorkomen zijn de ziekte van
Gaucher, de metachromatische leukodystrofie en de ziekte van
Pompe.
Het is niet mogelijk om hier in te gaan op de symptomatologie van
de diverse lysosomale stapelingsziekten. Hiervoor kan worden
verwezen naar onder andere Scriver et al (1), Fernandes et al (2)
en Siegel et al (3). Tabel 1 geeft aan in welke gevallen gedacht
moet worden aan een lysosomaal defect (4).

In de loop der jaren is een groot aantal lysosomale
enzymdeficiënties beschreven die ingedeeld kunnen worden in
groepen op basis van het type van verbindingen waarvan de
afbraak is verstoord: sfingolipidosen, mucopolysaccharidosen en
oligosaccharidosen.
Voor transport van de monomoleculaire componenten na afbraak
door de katabole enzymen zijn transporteiwitten in de membraan
verantwoordelijk. Ook hier zijn deficiënties bekend waardoor
stapeling van neuraminezuur of cystine kan optreden.
Bovengenoemde defecten worden veroorzaakt door mutaties in de
katabole enzymsystemen of in de transporteiwitten zelf.
Het defect kan ook op een hoger niveau liggen waarbij door een
enzymdeficiëntie in het Golgi systeem de routing van een hele
groep van lysosomale enzymen verstoord is en er een deficiëntie
van een groep van lysosomale enzymen ontstaat. Dit is het geval
bij de mucolipidosen II en III.
Op basis van de kliniek en het waargenomen stapelingsmateriaal
in weefsels en lichaamsvloeistoffen is de indeling van
verschillende groepen ontstaan: sfingolipidosen,
oligosaccharidosen en mucopolysaccharidosen.
Men moet zich echter realiseren dat vaak de specificiteit
van een enzym zich beperkt tot de af te splitsen groep, wat
betekent dat één enzym betrokken kan zijn bij de afbraak van
verschillende groepen van verbindingen. Zo bevatten zowel
glycoproteïnen als een deel van de sfingolipiden oligosaccharide
ketens waarvan de afbraak voor een deel door dezelfde enzymen
wordt gekatalyseerd. In deze gevallen ontstaat stapeling niet
alleen van sfingolipiden maar ook van oligosacchariden afkomstig
van glycoproteïnen, waarbij de klinische consequenties van de
sfingolipide stapeling op de voorgrond staan en de meeste van
deze defecten daarom tot de sfingolipidosen worden gerekend.
Sfingolipidosen:
De sfingolipidosen (tabel 2) vormen een groep van lysosomale
stapelingsziekten waarbij sprake is van een genetisch defect in
één van de enzymen betrokken bij de afbraak van de
sfingolipiden.
Terwijl bij de fosfolipiden en triglyceriden het glycerolskelet de
basisstructuur van het molecuul vormt, is dit bij de sfingolipiden
het sfingosine molecuul, structureel vergelijkbaar met een
monoglyceride. De aminogroep van het sfingosine is geacyleerd met
een vetzuur.
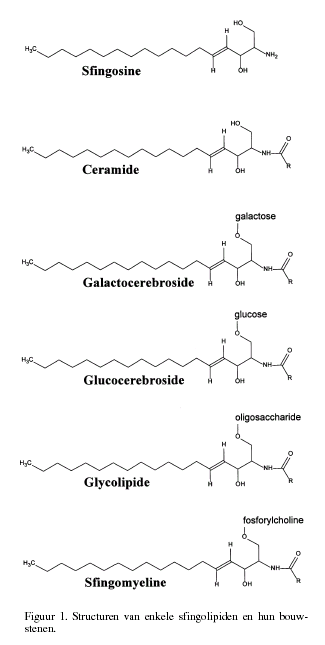
Ceramide is qua structuur vergelijkbaar met een diglyceride (fig.
1). Aan de hydroxylgroep van het ceramide op de 1- plaats kunnen
diverse groepen gekoppeld zijn. Als de hydroxylgroep veresterd is
met fosforylcholine ontstaat het fosfolipide sfingomyeline. Bij de
glycolipiden is de hydroxylgroep geglycosyleerd met een suiker
(cerebroside) of een oligosaccharide. Glycolipiden vormen vooral
in de hersenen een belangrijke groep van verbindingen.
De voornaamste samenstellende lipiden in de hersenen zijn
galactocerebroside en sulfatide (op de 3’-plaats gesulfateerd
galactocerebroside). Deze cerebrosides vormen samen met
cholesterol de hoofdcomponenten van de witte stof met name van het
myeline.
Hersenweefsel bevat daarnaast een groot aantal glycolipiden met
meer dan één suikerresidu. Ze zijn afgeleid van glucocerebroside.
De samenstellende suikers zijn galactose (Gal), glucose (Glc),
N-acetylglucosamine (GlcNac), N-acetylgalactosamine (GalNac),
fucose (Fuc) en N-acetyl-neuraminezuur (NANA) (5).
Een belangrijke groep van deze glycolipiden met complexe
suikerstructuur vormen de gangliosiden gekenmerkt door de
aanwezigheid van één of meer neuraminezuur suikers in de
suikerketen. Door de aanwezigheid van een carboxylgroep in het
neuraminezuur gedragen de gangliosiden zich als zure moleculen.
Een veel gebruikte nomenclatuur voor de gangliosiden is die van
Svennerholm. Zijn classificatie is gebaseerd op het aantal
neuraminezuur groepen aanwezig in het molecuul en de loopsnelheid
op TLC (5).
GM1- en G-gangliosiden zijn monosialyl (M)verbindingen en bevatten
één neuraminezuurgroep. GM1 is opgebouwd uit een suikerketen van 4
suikerswaarbij neuraminezuur als vertakking aanwezig is:
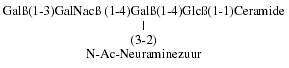
Bij GM2 is de eindstandige galactosegroep afgesplitst.
Omdat sfingolipiden belangrijke bestanddelen vormen van het
perifeer en centraal zenuwstelsel gaat een defect in de afbraak
ervan bij vele van deze ziekten gepaard met neurologische
aandoeningen. De sfingolipiden worden afgebroken door een
achtereenvolgende afsplitsing van eindstandige moleculen van het
hydrofiele of polaire deel van het molecuul.
Dit kunnen suikermoleculen (glycolipiden), sulfaationen
(sulfatiden) of fosforylcholine (sfingomyeline) zijn.
Van het uiteindelijke resulterende ceramide skelet wordt het
vetzuur, gebonden aan de aminegroep van het sfingosine
afgesplitst door het ceramidase.
Als op deze manier de glycosidische, de ester- en de N-acyl -
bindingen verbroken zijn, worden de gevormde monomoleculaire
componenten over de lysosomale membraan getransporteerd en zijn
voor verder metabolisme beschikbaar.
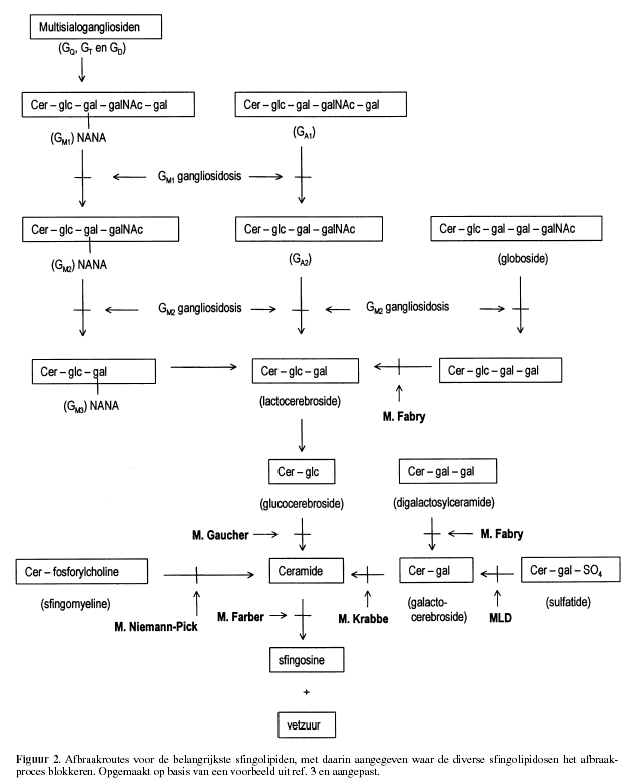
Deficiënties zijn bekend voor vrijwel alle enzymen betrokken bij
de afbraak van de sfingolipiden (fig. 2). Voor de afbraak van de
hydrofobe sfingolipiden met korte hydrofiele groepen worden
kleine nonenzymatische glycoproteïnen als activatoren gebruikt:
sfingolipid activator proteins (SAPS): GM2 activator protein en
de groep van SAP A, B, C, en D proteïnen. Ook van deze eiwitten
zijn deficiënties bekend, aanleiding gevend tot een klinisch
beeld dat vergelijkbaar is met wat past bij een deficiëntie van
het enzym, dat onder normale omstandigheden door de activator
geactiveerd zou worden.

Lipidosen:
De hydrolyse van de vetzuur ester binding in triglyceriden en
cholesterolester in verband met de afbraak van triglyceriden en
cholesterolesters wordt verzorgd door het enzym zure lipase.
Een deficiëntie van dit enzym resulteert in accumulatie van
cholesterolesters en triglyceriden in de weefsels.
Afhankelijk van de leeftijd waarop de ziekte zich openbaart en
de ernst van de klinische symptomen worden hier twee fenotypen
onderscheiden.
De ziekte van Wolman is de ernstige, infantiele vorm.
De cholesterol ester storage disease (CESD) is veel milder en
wordt vaak pas op volwassen leeftijd gediagnostiseerd.
Niemann-Pick C is een lipidose, die veroorzaakt wordt door een
defect in het intracellulaire transport van exogeen cholesterol,
resulterend in een accumulatie van cholesterol in het lysosoom.
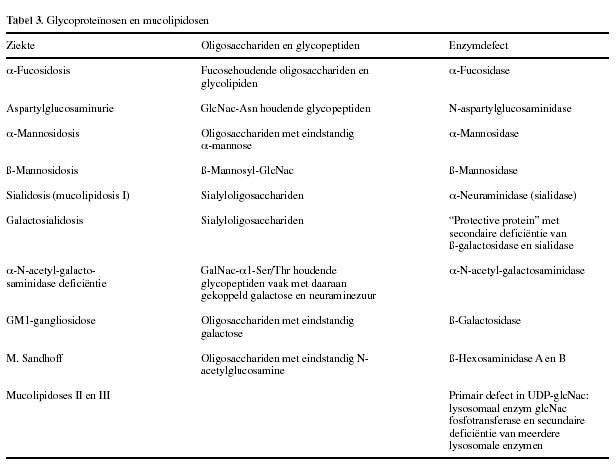
Oligosaccharidosen / Glycoproteïnosen:
Onder de groep van oligosaccharidosen wordt verstaan de lysosomale
stapelingsziekten met oligosacchariden als belangrijkste
stapelingsproduct. De oligosacchariden zijn afkomstig van
glycoproteïnen. Een andere naam voor deze groep is die van de
glycoproteïnosen (tabel 3).
Glycoproteïnen kunnen Nglycosidische, aan asparagine (Asn), of
O-glycosidische, aan serine (Ser) of threonine (Thre) gekoppelde
oligosacchariden bevatten.
De N-glycosidische oligosacchariden zijn van het complexe,
oligomannose, of hybride type en worden opgebouwd via een
ingewikkelde synthese route die elders in deze editie van het
tijdschrift wordt behandeld.
De afbraak van glycoproteïnen in lysosomen wordt wat betreft het
eiwitdeel verzorgd door proteasen. Het oligosaccharide wordt tot
losse suikers afgebroken door de glycosidasen.
N-aspartylglucosaminidase verbreekt bij de Nglycosidisch gebonden
oligosacchariden de binding met het asparagine (fig. 3a en 3b).
Een endoglycosidase splitst de binding tussen de twee
N-acetylglucosamine moleculen. Het oligosaccharide wordt verder
net als bij de glycolipiden door exoglycosidasen afgebroken.
Figuur 3 illustreert welke enzymen betrokken zijn bij de afbraak
van de verschillende typen oligosacchariden.
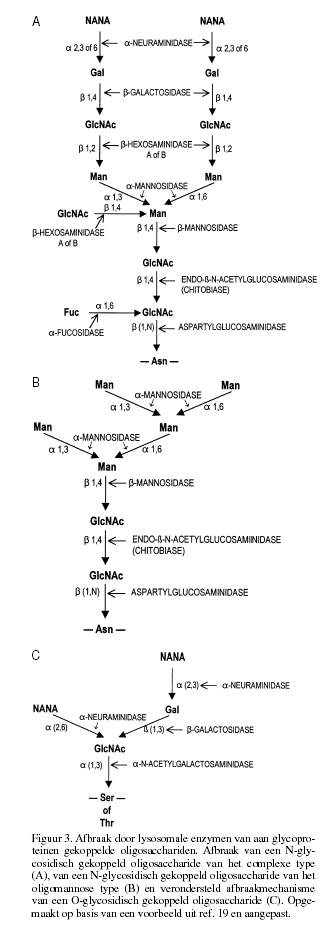
Een deficiëntie van één van de enzymen heeft tot gevolg dat de
rest van de oligosaccharideketen zich stapelt.
De verschillende oligosaccharidosen geven elk een karakteristiek
patroon van oligosacchariden. De O-glycosidisch gebonden
oligosacchariden (fig. 3c) zijn opgebouwd uit
N-acetylgalactosamine, galactose, N-acetylglucosamine, fucose en
neuraminezuur. De synthese vindt plaats via directe overdracht van
aan nucleotiden gekoppelde suikers aan de suikerketen. Hierbij is
een grote diversiteit in structuren mogelijk.
Over de afbraak van O-glycosidisch gekoppelde suikers is in
vergelijking met de N-glycosidische minder bekend.
De meeste oligosacchariden beschreven bij lysosomale
stapelingsziekten zijn van het N-glycosidische type
a-Nacetylgalactosamine is een belangrijke bouwsteen van
O-glycosidisch aan glycoproteïnen gebonden oligosacchariden.
De basisstructuur bij de O-glycosidisch gebonden oligosacchariden
kunnen in twee klassen worden ingedeeld: Galß(1-3)GalNac-a-Ser (of Thr) en GlcNac-ß(1-3)-GalNac-a-Ser (of Thr). Beide klassen bevatten dus
a-GalNac-residuen gekoppeld
via serine of threonine aan de eiwitketen (6). Enkele van de
bovengenoemde enzymen zijn ook actief op glycolipiden en worden
daarom ook vaak ingedeeld bij de sfingolipidose enzymen:
ß-N-Acetylhexosaminidase, ß-galactosidase.
Behalve oligosaccharide uitscheiding wordt bij deficiënties van
deze enzymen ook stapeling van sfingolipiden waargenomen. Zij
worden ingedeeld bij de sfingolipidosen omdat de
sfingolipidestapeling op de voorgrond staat en ze zich ook
klinisch van de echte glycoproteïnosen onderscheiden.
Glycogenosen:
De glucose moleculen van glycogeen in het lysosoom worden
afgesplitst door het a-glucosidase (zure
maltase). Bij een deficiëntie van dit enzym (de ziekte van Pompe)
accumuleert glycogeen in de weefsels.
De ziekte van Pompe wordt ook tot de glycogenosen (type II)
gerekend. Afbraakproducten van het accumulerende glycogeen worden
bij M. Pompe uitgescheiden als een tetraglucoside-oligosaccharide
samen met hogere polymeren van glucose (7).
Ook bij enkele andere glycogenosen kunnen deze oligosacchariden in
de urine worden aangetoond (type III, debrancher deficiency en
type IX, fosforylase kinase deficiency) indien er sprake is van
glycogeenstapeling (hepatomegalie) (8). Bij patiënten met late
onset vormen van deze ziekte is het tetraglucoside vaak niet
aantoonbaar in de urine zodat de diagnose met andere middelen,
zoals enzymdiagnostiek, gesteld zal moeten worden.
Mucolipidose II en III / I-cell disease:
Bij de mucolipidosen II en III (tabel 3) wordt door een post
translatiedefect in de biosynthese van de oligosaccharide keten
van een aantal lysosomale enzymen de routing van de enzymen naar
het lysosoom verstoord.
De enzymen worden niet naar het lysosoom getransporteerd maar
uitgescheiden door de cel. Dit leidt tot een deficiëntie van een
aantal lysosomale enzymen in de cel en verhoogde activiteiten in
plasma. De kliniek is ernstig. De diagnostiek berust op meting van
de enzymactiviteiten in plasma.
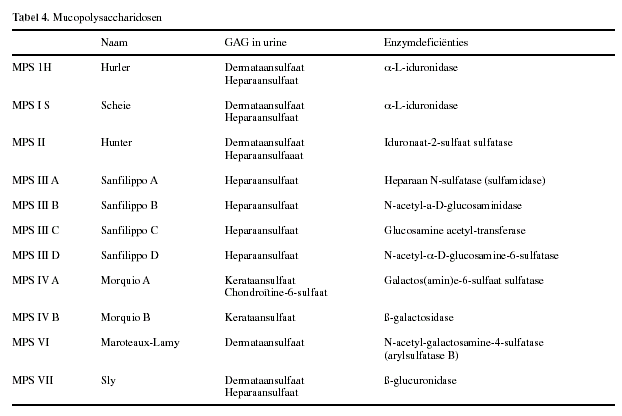
Mucopolysaccharidosen:
Bij patiënten met een mucopolysaccharidose (tabel 4) is één van de
lysosomale enzymen betrokken bij de afbraak van
glycosaminoglycanen GAG’s, (synoniem voor mucopolysacchariden),
deficiënt (9).
Glycosaminoglycanen zijn lange negatief geladen suikerketens,
opgebouwd uit repeterende disaccharide-eenheden. De
disaccharide-eenheid bepaalt het type GAG: chondroïtine sulfaat
(CS), dermataansulfaat (DS), kerataansulfaat (KS) of
heparaansulfaat (HS).
Bij alle GAG’s, behalve bij KS, is één van de samenstellende
suikers een uronzuur (glucuronzuur of iduronzuur). De suikerketens
kunnen op diverse plaatsen gesulfateerd zijn. De hexosamine van CS
en DS kan op de 4- of de 6-plaats, de hexose van KS op de 6-plaats
en het uronzuur van CS, DS en HS op de 2-plaats veresterd zijn met
een sulfaatgroep. Bij HS kan naast de 6- OH-groep ook nog de
stikstofgroep van de hexosamine gesulfateerd zijn.
In de weefsels en cellen zijn GAG’s gebonden aan een eiwitketen
("core-protein") en worden in die vorm proteoglycanen
genoemd. De binding aan de eiwitketen is veelal een binding aan
een serinemolecuul via een xylose-galactose-galactose
trisaccharide. Op die manier kunnen er één of meer GAG-moleculen
van hetzelfde type of van verschillende typen aan het core
proteïne gekoppeld zijn. GAG’s en proteoglycanen zijn voor het
merendeel mgelokaliseerd buiten de cel.
De afbraak van de GAG’s tot monomoleculaire componenten door
proteasen, sulfatasen en glycosidasen vindt intracellulair plaats
in de lysosomen.
Extracelulaire protease- en endoglycosidase-activiteiten kunnen
echter ook een rol spelen in het katabolisme van de
proteoglycanen. Om volledig afgebroken te kunnen worden, moeten de
proteoglycanen of GAG’s door middel van endocytose over de
celmembraan naar de lysosomen getransporteerd worden.
Endoglycosidasen, zoals hyaluronidase, heparanoglucuronidase en
endo-ßgalactosidse, zijn in staat om ergens midden in de GAG-keten
de glycosidische binding te splitsen. Op die manier worden meer
eindstandige suikergroepen gecreëerd en de substraatconcentratie
voor de exoenzymen verhoogd.
De splitsing van de GAG keten in monomoleculaire componenten
(sulfaationen en suikermoleculen) wordt verzorgd door een team van
exosulfatasen en exoglycosidasen. De volgorde van de afbraak ligt
vast omdat de exo-enzymen alleen eindstandige sulfaatgroepen en
suikers afsplitsen. Een deficiëntie van een exo-enzym heeft
stapeling van de rest van de GAG-keten tot gevolg.
Neuronale Ceroid Lipofuscinoses (NCL):
De Neuronale Ceroid Lipofuscinoses (NCL’s) vormen een groep van
ernstige neurodegeneratieve aandoeningen gekenmerkt door een
stapeling van autofluorescerend materiaal, voornamelijk in het
centrale zenuwstelsel (10).
Klinisch worden de NCL’s gekenmerkt door onder andere progressief
verlies van gezichtsvermogen, uiteindelijk leidend tot blindheid,
vaak therapie resistente epileptische aanvallen, verlies van
motore en intellectuele functies en vroegtijdig
overlijden.
Reeds lang bestond het vermoeden dat het hier om lysosomale
stapelingsziekten ging, omdat met markers kon worden aangetoond,
dat het stapelingsmateriaal gelokaliseerd was in lysosomale
structuren.
De NCL’s voldeden echter niet aan de klassieke definitie van
lysosomale stapelingsziekten, omdat tot voor kort er geen
lysosomaal (enzym)defect bekend was.
Op basis van de resultaten van recent moleculair genetisch
onderzoek zijn tenminste 8 verschillende NCL genen
(CNL1-8) betrokken bij dit ziektebeeld. Dit heeft geleid
tot een herclassificering van de NCL-ziekten.
Deze is nu gebaseerd op het onderliggend gen defect.
Voor een aantal is het gen product en de functie ervan bekend. Op
basis hiervan kunnen enkele reeds met zekerheid tot de groep van
lysosomale stapelingsziekten gerekend worden.
Voor twee van de genen (CLN1 en CLN2) is aangetoond dat ze
coderen voor lysosomale enzymen.
Drie andere
(CLN3, 5 en 8)
coderen voor transmembraan eiwitten, waarvan de functies nog
onbekend zijn.
Voor CLN3 is lokalisatie in de lysosomale membraan
aangetoond.
Het CLN1 gen codeert voor palmitoylprotein thioesterase
(PPT) en defecten in dit enzym leiden tot de infantiele vorm van
NCL (M. Santavuori- Haltia).
PPT is een lysomaal enzym dat palmitoyl thioesters, van met
vetzuur geacyleerde cysteïne residuen in
eiwitten, deacyleert.
Het gen product van het CLN2 gen is tripeptidyl-peptidase
I. Deficienties van dit enzym zijn de oorzaak van de klassieke
laat infantiele vorm van NCL (M. Jansky-Bielschowsky).
Het CLN3 gen, wat codeert voor een lysosomaal membraan
eiwit, is betrokken bij het klassieke juveniele type (M. Batten).
Omdat ook de variant laat infantiele NCL’s, waaraan defecten in de
CLN5, 6 en 7 genen ten grondslag liggen, zowel qua kliniek als
pathologie verwant zijn aan de rest, is het waarschijnlijk dat het
ook bij deze types om defecten in lysosomale eiwitten gaat.
Het gen voor de adulte vorm (Kufs disease) is nog niet bekend en
mogelijk is bij dit ziektebeeld meer dan één gen betrokken. De
eiwitdiagnostiek voor CLN1 en CLN2 is nu op het
zelfde niveau van de andere bekende lysosomale stapelingsziekten
(11-13).
Transportdefecten:
Na afbraak van verbindingen als triglyceriden, fosfolipiden,
sfingolipiden, glycoproteïnen en glycosaminoglycanen resteren de
moleculaire bouwstenen zoals vetzuren, glycerol, fosfaat,
ceramide, sulfaat, monosacchariden en aminozuren.
Het transport hiervan over de lysosomale membraan naar het
cytoplasma vindt plaats veelal via specifieke transportmoleculen
in de lysosomale membraan. Ook hier zijn defecten bekend, die
aanleiding geven tot stapeling van deze monomoleculaire
componenten.
Het defect in transport van neuraminezuur is de meest bekende,
aanleiding gevend tot (sterk) verhoogde uitscheiding van
neuraminezuur.
Screening op stapelingsproducten in urine:
Door het defect in de lysosomale afbraak zullen één of meer
producten zich stapelen in het lysosoom. Secundair daaraan vindt
vaak ook nog ophoping van andere niet direct aan het enzymdefect
gerelateerde verbindingen plaats, waarschijnlijk doordat ook de
andere niet deficiënte enzymen in hun functie belemmerd worden
door het stapelingsfenomeen en de abnormale lysosomale inhoud en
structuur (14).
Door de stapeling groeien de lysosomen uit tot vacuoolachtige
structuren, die microscopisch (licht en EM) herkenbaar zijn.
Morfologisch onderzoek van weefsel, leukocyten en beenmerg kan
vaak een belangrijke indicatie geven voor het aanwezig zijn van
een lysosomaal stapelingsfenomeen.
Door celdood kunnen de stapelingsproducten, afhankelijk van hun
oplosbaarheid, in de circulatie komen en via de urine worden
uitgescheiden (oligosacchariden, mucopolysacchariden en
neuraminezuur).
Door het aantonen van verhogingen in de concentratie van deze
producten in urine kan worden voorgescreend op een aantal
lysosomale stapelingsziekten.
Oligosacchariden:
Oligosaccharide uitscheiding in urine wordt gezien bij de
glycoproteïnosen, een aantal sfingolipidosen en de ziekte van
Pompe.
In totaal kan door analyse van oligosacchariden in urine worden
voorgescreend op 10 verschillende lysosomale stapelingsziekten. De
meest gebruikte techniek voor de analyse van oligosacchariden is
één- dimensionale chromatografie op silicagel platen. Als
loopmiddel wordt gebruikt een mengsel van butanol, azijnzuur en
water in de verhouding 2:1:1.
Het urinemonster (portie) wordt van tevoren ontzout, omdat zouten
het loopgedrag van de oligosacchariden verstoren. Ontzouten kan
geschieden met behulp van ionenwisselingschromatografie, maar
eenvoudiger is een kleine hoeveelheid (100 mg) van een gemengde
ionenwisselaar, voorbehandeld met azijnzuur, aan het urinemonster
toe te voegen. Geladen oligosacchariden (glycopeptiden en
neuraminezuurhoudende oligosacchariden) kunnen hierbij echter
verloren gaan. Daarom wordt altijd naast het ontzoute monster ook
een niet ontzout monster gechromatografeerd.
De hoeveelheid opgebrachte urine is leeftijdsafhankelijk en er
wordt een correctie uitgevoerd voor het kreatinine gehalte van de
urine (15,16). Suikerhoudende banden worden specifiek gekleurd
door te sprayen met een orcinol oplossing in verdund zwavelzuur of
door de plaat te dompelen in een orcinol zwavelzuur oplossing in
aceton.
De oligosacchariden worden zichtbaar na verhitting.
Er bestaan, naast de voor suikers specifieke orcinolkleuring,
specifieke kleuringen voor neuraminezuur en neuraminezuur houdende
oligosacchariden (resorcinolkleuring) en glycopeptiden
(ninhydrinekleuring).
Om de positie van de oligosaccharide banden te kunnen bepalen
worden standaarden zoals glucose, lactose, raffinose,
tetraglucoside en sialyllactose in een mengsel meegenomen en een
monster van gedeeltelijk gehydrolyseerd dextraan, dat glucose
monomeer en de diverse
a-(1-6) dimeren van glucose
bevat (14).
Urines van jonge kinderen bevatten altijd een aantal normale
oligosacchariden, die onder andere afkomstig zijn van voeding.
Borstvoeding maar ook andere voedingen geven een patroon van
oligosaccharide banden, dat varieert met de voeding.
De banden in het oligosaccharide patroon kunnen worden
gekarakteriseerd door hun positie ten opzichte van die van de
standaarden en ten opzichte van de polymeren van glucose uit een
dextraanhydrolysaat (fig 4) te bepalen.
Het vereist ervaring om de normale (voedings) patronen te leren
kennen en te kunnen onderscheiden van de patronen, die passen bij
een lysosomaal defect.
Glycoproteïnosen
a-Fucosidosis
De afsplitsing van fucose moleculen gekoppeld aan
N-acetylglucosamine (GlucNac) residuen is een van de eerste
stappen in de afbraak van oligosacchariden. Dit omdat het volgende
enzym in de afbraakroute ( Naspartylglucosaminidase ) alleen
actief is als fucose, gekoppeld aan het aan asparagine gebonden
Gluc- Nac, is afgesplitst. Omdat het enzym aspartylglucosaminidase
geremd wordt door de aanwezigheid van een fucoseresidu aan de
N-acetylglucosaminegroep zullen bij een a-fucosidase deficiëntie
behalve fucosehoudende oligosacchariden, zoals Fuc-GlucNac, ook
fucoglycopeptiden, zoals Fuc-GlucNac–asp, worden uitgescheiden.
Daarnaast worden ook grotere glycopeptiden en oligosacchariden met
een fucoseresidu gekoppeld aan GlcNac verderop in de
oligosacchariden keten als stapelingsproduct waargenomen (17- 19).
Laan 7 in fig. 4 geeft het TLC patroon na orcinol kleuring.
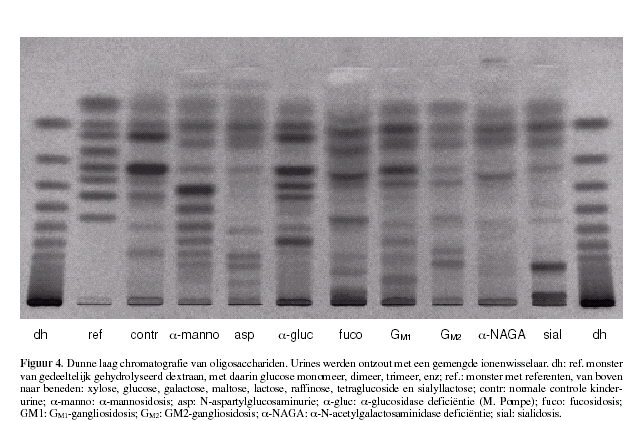
N-Aspartylglucosaminurie
Deficiëntie van het aspartylglucosaminidase verhindert de
splitsing tussen de N-acetylglucosamine suiker en het asparagine
van het glycopeptide, dat overblijft na de afbraak van de
eiwitketen door diverse proteolytische enzymen. De
oligosaccharideketen met twee N-acetylglucosamine residuen aan de
reducerende kant wordt kennelijk wel afgesplitst via een
endo-ß-N-acetylglucosaminidase (fig. 3a). Dit oligosaccharide
wordt door exoglucosidasen tot monosacchariden afgebroken.
Bij een deficiënte van het N-aspartylglucosaminidase resteert het
N-aspartylglucosamine.
Dit is het belangrijkste stapelingsproduct.
Het kan met behulp van dunne laag chromatografie in urine niet met
orcinol maar wel via de ninhydrinekleuring worden aangetoond.
Daarnaast zijn ook andere glycopeptiden in de urine aanwezig, die
wel via de normale screening op oligosacchariden met orcinol
kleuring kunnen worden gezien, als gevolgvan een gedeeltelijke
afbraak van de oligosaccharide keten, maar ook oligosacchariden
als galß-( 1-4) GlcNac-Asn eventueel nog uitgebreid met een
N-acetylneuraminyl- of N-acetylglucosaminyl-groep. Deze volgorde
van suikers komt normaal niet in Nglycosidisch gebonden
suikerketens voor. Ze worden klaarblijkelijk gevormd door
koppeling van suikers op basis van activiteiten van
galactosyltransferasen bij een overmatig aanbod van substraat
(17-19).
In het TLC patroon zijn 3-4 extra banden te zien na orcinol
kleuring (fig. 4 laan 5). De vrije –NHgroep van de glycopeptiden
kleurt met ninhydrine zodat bij de analyse van aminozuren in urine
2 afwijkende pieken gezien worden in het profiel van de aminozuren
afkomstig van N-aspartylglucosamine en gal-ß-(1-4) GlcNac-Asn.
a-Mannosidosis
Patiënten met een deficiëntie van a-mannosidase scheiden
MannGlcNac (n=2) oligosacchariden uit. Deze zijn afkomstig van de
N-glycosidisch gebonden oligosacchariden van het oligomannose type
gebonden aan de glycoproteïnen (fig. 3b). De urine bevat een groot
aantal van deze oligosacchariden waarvan Man2GlcNac in de hoogste
concentratie aanwezig is (fig. 4 laan 4). De hoeveelheid en
daarmee ook de intensiteit van de mannosyloligosacchariden neemt
af met toenemend aantal mannose residuen (7,20,21).
Sialidosis en galactosialidosis
Omdat neuraminezuur (sialic acid) bij de N-glycosidisch gebonden
oligosacchariden een eindstandige suiker is, zijn de
oligosacchariden uitgescheiden bij een neuraminidase deficiëntie
hogere oligosacchariden bestaande uit 6 of meer samenstellende
suikers. De belangrijkste oligosacchariden zijn volledig en
gedeeltelijk gesialyleerde mono- en disialyl-oligosacchariden.
Vrijwel dezelfde oligosacchariden worden gezien in een
galactosialidosis urine. De relatieve hoeveelheden van de
verschillende sialyloligosacchariden kunnen tot op zekere hoogte
variëren (24). In het oligosaccharidepatroon zijn twee hoofdbanden
duidelijk aanwezig (fig. 4 laan 11).
a-N-acetylgalactosaminidase deficiëntie
a-N-acetylgalactosamine is een belangrijke bouwsteen van
O-glycosidisch aan glycoproteïnen gebonden oligosacchariden. Het
komt ook in suikerketen van glycolipiden voor.
Stapelingsproducten afkomstig van O-glycosidisch gebonden
suikerketens worden gevonden in urine. Naar verwachting zullen
zich ook lipidstructuren stapelen.
De basisstructuur bij de O-glycosidisch gebonden oligosacchariden
kunnen in twee klassen worden ingedeeld: Galß(1-3)GalNac-Ser(of
Thr) en GlcNac-ß(1-3)-GalNac-Ser(of Thr). Beide klassen bevatten
dus a-GalNac-residuen gekoppeld via serine of threonine aan de
eiwitketen. De stapelingsproducten gevonden in urine zijn als
gevolg hiervan glycopeptiden. De belangrijkste hiervan hebben
echter niet een a-N-acetylgalactosamine maar een neuraminezuur als
eindstandige suiker. De oorzaak hiervan is niet duidelijk.
Het oligosaccharide patroon laat drie afwijkende banden zien na
kleuring op neuraminezuur (resorcinol) waarvan één door de hogere
intensiteit duidelijk zichtbaar is na orcinol kleuring de andere
twee niet of kleuren erg zwak (fig. 4 laan 10). Kleuring op
neuraminezuur met behulp van resorcinol maakt kennelijk door de
hogere gevoeligheid alle drie de banden zichtbaar. Deze
neuraminezuur houdende glycopeptiden kunnen na
ontzoutingsprocedure met ionenwisselaars hieraan gebonden worden
en daardoor verdwijnen. Na ontzouten kan bij deze deficiëntie
zelfs een normaal patroon worden gevonden (16).
Sfingolipidosen
Belangrijkste componenten, waarvan het metabolisme verstoord is
bij de sfingolipidosen en lipidosen zijn de glycolipiden,
fosfolipiden, triglyceriden en cholesterolesters. Door hun slechte
wateroplosbaarheid worden ze niet in de urine teruggevonden (bij
een aantal sfingolipidosen, zoals bij de metachromatische
leukodystrofie en de ziekte van Fabry, wordt lipidestapeling
gezien in het urinesediment).
Bij enkele sfingolipidosen echter is het enzym ook betrokken bij
de afbraak van de oligosaccharideketen van de glycoproteïnen. Hier
wordt naast lipidestapeling ook stapeling van oligosacchariden
waargenomen. Ze kunnen daarom in principe ook tot de
glycoproteïnosen gerekend worden. Deze oligosacchariden worden
uitgescheiden in urine, waardoor screening mogelijk is.
GM1-gangliosidosis
GM1-gangliosidosis wordt veroorzaakt door een deficiëntie van het
ß-galactosidase. Omdat dit de eindstandige suiker is van het
GM1-ganglioside, een belangrijke component van neuronaal weefsel
met name de grijze stof, is GM1-ganglioside een belangrijk
stapelingsproduct.
ß-Glycosidisch gebonden galactose is echter ook één van de
samenstellende suikers in het complexe type oligosaccharide van de
glycoproteïnen (fig. 3a). Bij GM1-gangliosidosis wordt daarom ook
een karakteristiek afwijkend oligosaccharide patroon in de urine
waargenomen met 3-4 afwijkende oligosacchariden met ß-glycosidisch
aan de niet reducerende zijde gebonden galactose (fig. 4 laan 8)
(7,25-27).
GM2-gangliosidosis variant O (M. Sandhoff) Bij deze variant van de
GM2-gangliosidosis zijn beide iso-enzymen A en B van het
ß-N-acetylhexosaminidase deficiënt (28). Het B-iso-enzym heeft een
voorkeur voor neutrale wateroplosbare oligosacchariden (29).
Variant O (Morbus Sandhoff) wordt daarom gekenmerkt door stapeling
en excretie van oligosacchariden. Bij variant B (Tay-Sachs
disease) is alleen de hexosaminidase A activiteit deficiënt.
Hierbij wordt geen verhoogde uitscheiding van oligosacchariden
waargenomen. In het TLC patroon worden bij de ziekte van Sandhoff
twee karakteristieke banden waargenomen. Strecker et al (30) en
Warner et al (31) beschreven 10-12 N-acetylglucosamine bevattende
oligosacchariden bij Sandhoff patiënten met twee hoofdcomponenten,
waarschijnlijk corresponderend met de twee banden, die in het voor
M. Sandhoff karakteristieke TLC patroon zichtbaar zijn (fig. 4
laan 9).
Mucopolysacchariden
Minimaal 10 verschillende enzymen (glycosidasen en sulfatasen)
zijn nodig voor een volledige afbraak van de diverse GAG-ketens
tot de monomoleculaire componenten.
Omdat alleen eindstandige sulfaatgroepen of suikers afgesplitst
kunnen worden zal een deficiëntie van elk van deze enzymen
resulteren in een accumulatie van de rest van de GAG-keten in de
cellen en in de weefsels.
Op basis van deze enzymdeficiënties kunnen we 10 verschillende
erfelijke ziektebeelden onderscheiden, ieder met een eigen
stapelingsproduct of combinatie van stapelingsproducten.
Tabel 4 geeft een overzicht van de verschillende
mucopolysaccharidosen met de daarbij passende enzymdeficiënties
en de stapelingsproducten (9).
De verschillende mucopolysaccharidosen hebben gemeenschappelijke
klinische kenmerken. De meest kenmerkende hiervan zijn:
vergroting van de lever en/of milt ten gevolge van stapeling van
de GAG’s in deze weefsels, retardatie, grof gelaat,
(‘"gargoyle uiterlijk"), corneatroebeling, stijfheid
in de gewrichten en skeletafwijkingen. Voor een meer uitgebreide
beschrijving van de kliniek van de verschillende typen
mucopolysaccharidosen wordt verwezen naar Neufeld et al (32).
Screeningsprocedures
De GAG’s, die zich stapelen in de weefsels, kunnen door celdood
vrijkomen. Ze zijn goed wateroplosbaar en worden in de urine
teruggevonden.
Alle tot nu toe bekende mucopolysaccharidosen geven aanleiding tot
verhoogde uitscheiding van GAG’s in de urine (tabel 4). Meting van
GAG’s in urine is daarom algemeen gebruikt als screeningsprocedure
voor mucopolysaccharidosen. Een goede screeningsprocedure dient zo
weinig mogelijk vals negatieve uitslagen te geven (sensitiviteit)
moet gemakkelijke en snel uitvoerbaar zijn en een minimum aan vals
positieve uitslagen opleveren (specificiteit), omdat elke
positieve uitslag een vervolgonderzoek oplevert.
Screeningsprocedures voor mucopolysaccharidosen kunnen worden
onderverdeeld in kwalitatieve en kwantitatieve bepalingsmethoden.
De kwalitatieve testen zoals de spottest corrigeren over het
algemeen niet voor de urineconcentratie en kunnen daardoor vals
positieve en vals negatieve uitslagen geven.
De kwantitatieve testen kunnen worden uitgevoerd op een 24-uurs
urine, zodat de uitscheiding per 24-uur berekend kan worden.
Voor een screeningsprocedure heeft echter de bepaling in een
portie, waarbij gecorrigeerd wordt voor de urineconcentratie door
middel van het kreatinine gehalte de voorkeur. De
bepalingsmethoden voor GAG in urine meten het totaal aan GAG’s
(CS, DS, HS en KS).
De meeste testen maken gebruik van het feit dat GAG’s langgerekte
moleculen zijn met sterk negatief geladen sulfaatgroepen. Het
reagens is meestal een positief geladen (basisch) molecuul. De
resulterende complexen geven een troebeling (kationische
detergentia), meetbaar als verhoogde absorptie of
lichtverstrooiing, of een metachromatisch effect (basische
kleurstoffen), meetbaar als een verhoging van de absorptie bij een
bepaalde golflengte.
Andere testen meten het uronzuurgehalte na precipitatie
(carbazoltest). Van genoemde testen zijn de carbazoltest en de
kwantitatieve metachromatische testen het meest betrouwbaar.
Uronzuur carbazoltest
Deze test meet het uronzuur (glucuronzuur en iduronzuur) gehalte
in GAG, geprecipiteerd in een kationisch detergens (CPC, CETAB).
De test is betrouwbaar maar bewerkelijk. Kerataansulfaat het
uitscheidingproduct bij de ziekte van Morquio bevat geen
uronzuurresiduen en wordt daarom niet meegemeten. Patiënten met M.
Morquio type A hebben echte naast een verhoging van het
kerataansulfaat ook een verhoging van het chondroitine-6-sulfaat
gehalte in de urine, zodat met deze test toch een afwijkend
verhoogde waarde in de urine zal worden gemeten.
Metachromatische testen
Metachromasie treedt op door de reactie van de basische
kleurstoffen (toluidine blauw, Alcian Blue, Azure A,
dimethylmethyleen blauw). De metachromasie treedt op door de
regelmatige rangschikking van de kleurstofmoleculen langs de
rechte polyanionische matrix (33).
De spottesten zijn kwalitatieve metachromatische testen, die
niet corrigeren voor de mate van geconcentreerdheid van de urine.
Uit onderzoek is gebleken dat de spottesten niet betrouwbaar zijn.
Zowel vals positieve uitslagen als vals negatieve uitslagen werden
in deze studies gezien. De Ames spottest bijvoorbeeld, gebaseerd
op de metachromatische kleurstof Azure A, gaf in een serie van 75
urines met 26 mucopolysaccharidosis urines, getest door twee
verschillende laboratoria respectievelijk 19 en 35% vals negatieve
en 22 en 12% vals positieve uitslagen.
Met de kwantitatieve dimethylmethyleen blauw (DMB) methode (34)
werden in deze serie geen vals negatieve en geen vals positieve
uitslagen gevonden. De spottesten zijn dus onbetrouwbaar en
gebruik ervan als screeningsprocedure voor mucopolysaccharidosen
is niet meer verantwoord.
Door ons is een screeningsprocedure ontwikkeld op basis van de
dimethylmethyleen blauw (DMB), waarmee direct in urine dus zonder
voorbewerking van het monster, gemeten kan worden (34). De methode
gaf een goede correlatie met uronzuur-carbazoltest. De screening
kan worden uitgevoerd in urineporties, waarbij gecorrigeerd wordt
voor de geconcentreerdheid van de urine door de gemeten waarden
uit te drukken per mmol kreatinine. De referentiewaarden blijken
sterk leeftijdsafhankelijk te zijn en vooral in het eerste
levensjaar sterk af te nemen met de leeftijd. De voor de
verschillende leeftijdscategorieën vastgestelde referentiewaarden
bewegen zich binnen betrekkelijk nauwe grenzen.
Omdat de oorspronkelijke DMB methode gestoord wordt door eiwit in
de urine werd een gewijzigde DMB-bepaling ontwikkeld. Door
toevoeging van Tris-base aan de bepaling kan ook in aanwezigheid
van relatief hoge concentraties van eiwit (uitgetest tot een
concentratie van 5 gram per liter) het GAG-gehalte betrouwbaar
gemeten worden (35).
Belangrijk bij het GAG assay is de keuze van het
dimethylmethyleenblauw. De kwaliteit ervan kan worden afgelezen
aan het spectrum. De absorptie bij de golflengte van het DMB-GAG
complex moet in de DMB oplossing zonder GAG voldoende laag zijn om
een toename van de absorptie goed te kunnen meten. Daarnaast moet
het gehalte aan DMB in het preparaat en daarmee in de oplossing,
die hiermee gemaakt wordt, voldoende hoog zijn, om al het GAG in
het monster te kunnen binden. Een maat hiervoor is de absorptie
van de DMB oplossing. De ijklijn van de bepaling heeft een
beperkte lineariteit. Omdat GAG concentraties door variatie in
kreatinine gehalte van de urines en variatie in leeftijd van de
patiënten sterk kunnen uiteenlopen, moet altijd nagegaan worden of
de gemeten waarde in het lineaire gebied van de ijklijn ligt en of
het monster verdund moet worden ingezet.
Zeker bij mucopolysaccharidose patiënten zal de waarde al gauw in
het niet lineaire gebied liggen, waardoor een te lage waarde wordt
gemeten en patiënten gemist kunnen worden. Indien tijdens de
screening op mucopolysaccharidosen een verhoogdewaarde voor het
mucopolysaccharidegehalte wordt gevonden, volgt hierop een analyse
van de samenstelling van de glycosaminoglycanen met behulp van
1-dimensionale elektroforese (36). Tevens worden in leukocyten,
geïsoleerd uit heparinebloed de verschillende enzymen betrokken
bij het metabolisme van GAG’s gemeten.
Normale urine bevat voornamelijk chondroïtinesulfaat. Bij een
mucopolysaccharidose wordt na 1-dimensionale elektroforese een
afwijkende samenstelling gevonden. Verhogingen van GAG zonder dat
hieraan een erfelijke stofwisselingsziekte ten grondslag ligt
worden onder andere gezien bij patiënten met reumatische ziekten,
na myocard infarct en heparinetherapie. Ook zijn artificieel
verhoogde uitslagen mogelijk door verontreiniging van de urine met
lijmstoffen afkomstig van de rand van de urine opvangzakjes. Voor
een uitgebreider overzicht zie Blau et al (37).
Neuraminezuur:
Infantile sialic acid storage disease (ISSD) en Salla disease.
Defecten in transport van neuraminezuur kunnen worden opgespoord
door neuraminezuur analyse in urine. Dit is in principe mogelijk
door middel van resorcinol kleuring van de neuraminezuur band na
dunne laag chromatografie.
Omdat de verhogingen met name bij Salla disease gering kunnen zijn
heeft een kwantitatieve meting van het neuraminezuur, gecorrigeerd
wordt voor het kreatininegehalte van de urine, de voorkeur.
Neuraminezuur kan kwantitatief gemeten worden met behulp van HPLC
of enzymatisch, waarbij het na omzetting door
N-acetyl-neuraminezuur aldolase als pyruvaat wordt gemeten. Meting
van glycosaminoglycaan gehalte in urine is een goede voorscreening
voor de gehele groep van de mucopolysaccharidosen.
Bij de mildere vormen van de mucopolysaccharidosen kunnen de
verhogingen gering zijn en is een nauwkeurige meting vereist.
Adulte patiënten met de ziekte van Morquio kunnen een normaal
glycosaminoglycaangehalte in de urine hebben. Volwassen Morquio
patiënten zijn echter op basis van het klinisch beeld goed te
herkennen.
Analyse van oligosacchariden is een goede voorscreening voor de
groep van de oligosaccharidosen en een deel van de
sfingolipidosen, hoewel in een enkel geval de patronen moeilijk
zijn te interpreteren en soms, met name bij de milde vormen, de
afwijkingen moeilijk zijn vast te stellen. Bij gevonden
afwijkingen in het oligosaccharide-patroon, niet te verklaren op
basis van voeding of medicatie en bij gevonden verhoging van de
glycosaminoglycaan uitscheiding, wordt lysosomaal enzymonderzoek
geadviseerd.
Voor een aantal lysosomale stapelingsziekten met name uit de groep
van de sfingolipidosen en mucolipidosen is geen voorscreening
mogelijk. Dit betekent dat vaak rechtstreeks een beroep zal moeten
worden gedaan op enzymbepalingen om een lysosomaal defect te
bevestigen dan wel uit te sluiten.
Enzymdiagnostiek:
Verreweg het grootste deel van de lysosomale enzymdefecten kan
betrouwbaar in leukocyten worden vastgesteld. De leukocyt bevat
een vrijwel compleet arsenaal aan lysosomale enzymen en is daarmee
bij uitstek geschikt voor de diagnostiek van lysosomale
enzymdefecten (4).
Via dextraansedimentatie wordt uit een buis heparinebloed (10-15
ml, bij jonge kinderen minimaal 5-6 ml) een hoeveelheid leukocyten
geïsoleerd, voldoende om een tiental lysosomale enzymen
kwantitatief te meten. Ook na verzending per post expresse zodat
het materiaal de volgende morgen arriveert kunnen nog betrouwbare
enzymspiegels in het bloedmonster worden gemeten (4).
Voor een goede en zo volledig mogelijke lysosomale diagnostiek is
naast het bloedmonster echter ook een urinemonster nodig, omdat
hierin al reeds een aantal lysosomale ziekten kunnen worden
uitgesloten, waardoor het bloedmonster zo economisch mogelijk kan
worden gebruikt voor het uitsluiten van de resterende lysosomale
stapelingsziekten.
Oligosaccharidosen en mucopolysaccharidosen:
Indien na voorscreening in urine afwijkingen zijn gezien in het
oligosaccharide patroon of in het mucopolysaccharide gehalte en/of
de samenstelling wordt het enzymdefect enzymatisch bevestigd of
uitgesloten door meting van de enzymen in een bloedmonster.
Bij de voorscreening op oligosaccharidosen en
mucopolysaccharidosen moet men zich realiseren, dat in een aantal
gevallen, met name bij de milde vormen, de afwijkingen in het
patroon respectievelijk de concentratie gering kunnen zijn en
moeilijk vast te stellen.
Sfingolipidosen en lipidosen:
Het merendeel van de defecten in de groep van de sfingolipidosen
en de groep van de lipidosen resulteert in stapeling van alleen
lipiden zonder oligosacchariden (tabel 2). Hierdoor is een
voorscreening in urine op deze defecten door middel van
oligosaccharide analyse niet mogelijk.
Dit betekent dat hier rechtstreeks een beroep zal moeten worden
gedaan op enzymbepalingen om een lysosomaal defect te bevestigen
dan wel uit te sluiten.
Neuronale Ceroid Lipofuscinosen:
Ook voor de groep van de Neuronale Ceroid Lipofuscinosen (NCL’s)
is geen voorscreening in urine bekend.
Voor de infantiele (M. Santavuori-Haltia) en de klassieke laat
infantiele vorm (M. Jansky-Bielschowsky) van de NCL’s, veroorzaakt
door defecten in respectievelijk het CNL-1 en het
CNL-2 gen is de eiwitdiagnostiek nu op hetzelfde niveau van
de andere bekende lysosomale stapelingsziekten.
Voor de infantiele vorm is sinds kort een fluorometrisch substraat
beschikbaar voor meting van de activiteit van het
palmitoyl-protein thioesterase wat geschikt is voor pre- en
postnatale enzymdiagnostiek (11).
Voor de laat infantiele vorm (CLN2) is eveneens een
fluorimetrisch substraat beschikbaar voor de bepaling tripeptidyl
peptidase I (12 en 13).
Voor de andere NCL types is geen eiwit diagnostiek beschikbaar.
Overige:
Enkele lysosomopathieën kunnen alleen in plasma worden aangetoond
(mucolipidose II en III).
In de zeldzame gevallen waar het enzymdefect moeilijk of
niet in leukocyten kan worden aangetoond, kan het vrijwel altijd
in gekweekte huidcellen, fibroblasten, worden gemeten.
De ziekte van Niemann-Pick C, een defect in de verwerking van
cholesterol in de cel, aanleiding gevend tot stapeling van
cholesterol in lysosomen, kan alleen in een fibroblasten-kweek
worden aangetoond door middel van een fillipinekleuring op het
gestapelde cholesterol in de lysosomen van de fibroblasten.
Voor de diverse analyses zijn in de meeste gevallen op commerciële
basis de noodzakelijke substraten verkrijgbaar. Meestal gaat het
om fluorescentie metingen.
Een representatief deel van het natuurlijk substraat van het enzym
is gekoppeld aan het 4-methylumbelliferon (4-MU). In deze
gekoppelde toestand fluoresceert het methylumbelliferon niet. Na
incubatie van het leukocyten materiaal, bij het voor de lysosomale
enzymen zure pH optimum, wordt door het te meten enzym het deel
van het substraat, dat zijn specificiteit bepaalt afgekoppeld van
het 4-MU. Het 4 -MU fluoresceert als vrij molecuul na verhoging
van de pH.
Dit betekent dat na de enzymreactie het reactiemengsel basisch
moet worden gemaakt.
Voor een aantal van de lysosomale enzymen kan het substraat
niet op deze manier, dat wil zeggen met een artificieel substraat,
worden nagebootst, omdat het substraat op die manier niet
specifiek genoeg is en ook andere dan het te meten enzym met het
substraat kunnen reageren of omdat behalve de af te splitsen groep
ook de rest van het molecuul van belang is voor de activiteit van
het enzym. In die gevallen wordt een substraat gebruikt, dat qua
structuur het natuurlijk substraat zo veel mogelijk benadert. Vaak
is dit ook een fluorescerend of een radioactief gelabeld
substraat.
Voorbeelden van enzymen, waarvoor meer specifieke substraten
gebruikt worden zijn galactocerebrosidase en
sfingomyelinase.
De meeste lysosomopathieën erven autosomaal recessief over.
De ziekte van Fabry en de ziekte van Hunter zijn voorbeelden van
via het X-chromosoom overervende lysosomale ziekten.
De enzymatische activiteit bij homozygoten of hemizygoten voor het
gendefect is erg laag en varieert van 0 tot circa 10% van de
gemiddelde referentiewaarde.
Bij heterozygoten van de autosomaal recessief overervende
lysosomopathiëen wordt een restactiviteit van ongeveer 50%
gevonden en worden geen klinische symptomen waargenomen.
Voor de X-gebonden lysosomopathieën kunnen bij draagsters door
non-random inactivatie van het Xchromosoom wel klinische
verschijnselen optreden.
Pseudodeficiënties
Pseudodeficiënties worden gekenmerkt door een lage vaak haast
deficiënte activiteit van het gemeten lysosomale enzym, zonder dat
dit aantoonbare klinische consequenties heeft.
Een bekend voorbeeld is de pseudodeficiëntie bij de metachromatische
leukodystrofie. Twee mutaties, verantwoordelijk voor het merendeel
van de pseudodeficiëntie allelen, zijn in het arylsulfatase gen
aangetoond.
Bij homozygoten voor deze mutaties wordt een activiteit gemeten, die
5- 15% is van de normale activiteit en in de range ligt van
activiteiten die bij echte patiënten wordt gemeten. De frequentie
voor de pseudodeficiëntie mutaties, die meestal samen op hetzelfde
chromosoom voorkomen is 15-20%. Dit betekent dat zij in homozygote
vorm bij 2-4% van de populatie voorkomen (38). De mutaties zijn met
behulp van mutatie analyse makkelijk aan te tonen.
In een aantal gevallen, homozygotie voor het pseudodeficiëntie allel
bijvoorbeeld, kan echter na meting van de enzymactiviteit en
pseudodeficiëntie mutatie analyse niet vastgesteld worden of er
sprake is van homozygotie voor het MLD allel. In dat geval wordt bij
de verdenking op een metachromatische leukodystrofie een lipide
analyse op het urinesediment uitgevoerd en het gehalte aan
sulfatiden, het stapelingsmateriaal bij de MLD, gemeten. Bij MLD
patiënten is dit duidelijk verhoogd, bij pseudodeficiënten is het
sulfatide gehalte in het urinesediment niet hoger dan bij personen
met een normale activiteit. De lipide analyse geeft in deze gevallen
de doorslag voor de diagnostiek.
Chitotriosidase
Chitotriosidase is een plasma enzym afkomstig van geactiveerde
macrofagen. Het is een endo-ß-glucosaminidase. Chitotriosidase is
sterk verhoogd bij patiënten met de ziekte van Gaucher, maar ook
verhoogd in mindere mate bij 10 andere lysosomale
stapelingsziekten. Een verhoogde waarde wordt gemeten bij de
meerderheid van de patienten met de ziekte van Gaucher, Krabbe,
GM1-gangliosidose, Niemann-Pick A en B en Niemann-Pick C. De
verhoging is secundair aan het primaire defect bij deze
deficiënties en is opvallend hoog bij patiënten met de ziekte van
Gaucher. De verhoging van het chitotriosidase lijkt karakteristiek
voor lysosomale stapelingsziekten, omdat de verhoging niet wordt
gezien bij 20 nietlysosomale enzymdefecten of in plasma van
patiënten met hepatomegalie geassocieerd met infecties. Meting van
chitotriosidase in plasma kan een waardevolle aanvulling zijn op
de diagnostiek van lysosomale stapelingsziekten, omdat een
verhoging gemeten bij patiënten zonder diagnose indicatief is voor
een defect in het lysosomale proces (39).
Vervolgonderzoek:
Wordt een verlaagde enzym activiteit in leukocyten vastgesteld dan
wordt in verband met de ernst van de diagnose het onderzoek in
leukocyten herhaald en fibroblasten in kweek genomen om het defect
in een tweede celtype te kunnen bevestigen. Gekweekte huidcellen
worden bewaard voor eventueel DNA onderzoek en genetisch advies
binnen de familie.
Met dank aan:
Dr. O.P. van Diggelen voor zijn commentaar en aanvullingen met
betrekking tot de Neuronale Ceroid Lipofuscinosen, waardoor de
recente ontwikkelingen voor deze nieuwe groep van lysosomale
ziekten verwerkt konden worden.
Literatuur:
-
In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The
metabolic and molecular bases of inherited disease. New York:
McGraw-Hill, 1995: 2427-2879 en 3763-3797.
-
2. In: Fernandes J, Saudubray J-M, Van den Berghe G, eds.
Inborn metabolic diseases: diagnosis and treatment. Springer
Verlag, 1996: 375-395.
-
3. Suzuki K. Genetic disorders of lipid, glycoprotein, and
mucopolysaccharide metabolism. In: Siegel GJ, Agranoff BW,
Albers RW, Molinoff PB, eds. Basic neurochemistry. New York:
Raven Press, 1993: 793-813.
-
4. Wevers RA, Lamers KJB, Gabreels FJM. De diagnostiek van
lysosomale stapelingsziekten. Tijdschrift NVKC 1987; 12:
40-45.
-
5. Agranoff BW, Hajra AK. Lipids. In: Siegel GJ, Agranoff BW,
Albers RW, Molinoff PB, eds. Basic neurochemistry. New York:
Raven Press, 1993: 97-117.
-
6. Hirabayashi Y, Matsumoto Y, Matsumoto M. et al. Isolation
and characterization of major urinary amino acid O-glycosides
and a dipeptide O-glycoside from a new lysosomal storage
disorder (Kanzaki disease). J Biol Chem 1990; 265:
1693-1701.
-
7. Peelen GOH, de Jong JGN, Wevers RA. HPLC analysis of
oligosaccharides in urine from oligosaccharidosis patients.
Clin Chem 1994; 40: 914-921.
-
8. Kumlien J, Chester MA, Lindberg BS, Pizzo P, Zopf D,
Lundblad A. Urinary excretion of a glucose-containing
tetrasaccharide. A parameter for increased degradation of
glycogen. Clin Chim Acta 1988; 176: 39-48.
-
9. De Jong JGN, Wevers RA. Glycosaminoglycanen en
mucopolysaccharidosen. Tijdschr NVKC 1992: 17: 2-8.
-
Bennett MJ and Hofmann SL. J Inher Metab Dis 1999; 22:
535-544.
-
Diggelen OP van, Keulemans JL, Winchester B, Hofman IL,
Vanhanen SL, Santavuori P, Voznyi YV. Rapid fluorogenic
palmitoyl-protein thioesterase assay: pre- and postnatal
diagnosis of INCL. Mol Genet Metab 1999; 66: 240-4.
-
Vines DJ, Warburton MJ. Classicial late infantile
ceroilipofuscinosis fibroblasts are deficient in lysosomal
tripeptidylpeptidase I. FEBS Letters 1999; 443: 131-135.
-
Rawlings ND, Barrett AJ. Tripeptidyl-peptidase I is apparently
the CLN2 protein absent in classical late infantile neuronal
ceroid lipofuscinosis. Biochim Biophys Acta 1999; 1429:
496-500
-
De Jong JGN, Aerts JM, van Weely S, Hollak CE, van Pelt J, van
Woerkom LM, Liebrand-van Sambeek ML, Wevers RA.
Oligosaccharide excretion in adult Gaucher disease. J Inher
Metab Dis 1998; 21: 49-59.
-
Blom W, Luteyn JC, Kelholt-Dijkman HH, Huijmans JGM, Loonen
MCB. Thin-layer chromatography of oligosaccharides in urines
as a rapid indication for the diagnosis of lysosomal acid
maltase deficiency (Pompe’s disease). Clin Chim Acta 1983;
134: 221-227.
-
De Jong J, van den Berg C, Wijburg H, Willemsen R, van
Diggelen O, Schindler D, Hoevenaars F, Wevers R.
Alpha-N-acetylgalactosaminidase deficiency with mild clinical
manifestations and difficult biochemical diagnosis. J Pediatr
1994; 125: 385-391.
-
Warner TG, O’Brien JS. Genetic defects in glycoprotein
metabolism. Ann Rev Genet 1983; 17: 395-441.
-
Van Pelt J. Isolation and structural analysis of
sialyloligosaccharides from sialidosis and galactosialidosis
fibroblasts, placenta and urine. 1988. Thesis.
-
Thomas GH, Beaudet AL. Disorders of glycoprotein degradation
and structure: a-mannosidosis, ß-mannosidosis, fucosidosis,
sialidosis, aspartylglucosaminuria, and carbohydrate deficient
glycoprotein syndome. In: Scriver CR, Beaudet AL, Sly WS,
Valle D, eds. The metabolic and molecular bases of inherited
disease. New York: McGraw- Hill, 1995: 2529-2561.
-
Warner ThG, Mock AK, Nyhan WL, O’Brien JS. a-Mannosidosis:
analysis of urinary oligosaccharides with high performance
liquid chromatography and diagnosis of a case with unusually
mild presentation. Clin Gen 1984; 25: 248-255.
-
Lott IT, Daniel PP. Serum and urinary trisaccharides in
mannosisdosis. Neurology 1981; 31:1159-1162.
-
Dorland L, Duran M, Hoefnagels FET, Breg JN, Fabery de Jonge
H, Cransberg K et al. ß-Mannosidosis in two brother with
hearing loss. J Inher Metab Dis 1988; 11: 255-258.
-
Wenger DA, Sujansky E, Fennessey PV, Thompson JN. Human
ß-mannosidase deficiency. N Engl J Med 1986; 315:
1201-1205.
-
Van Pelt J, Kamerling JP, Bakker HD, Vliegenthart JF. A
comparative study of sialyloligosaccharides isolated from
sialidosis and galactosialidosis urine. J Inher Metab Dis
1991; 14: 730-740.
-
Suzuki Y, Sakuraba H, Oshima A. ß-Galactosidase deficiency
(ß-galactosidosis): GM1 gangliosidosis and Morquio B disease.
In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The
metabolic and molecular bases of inherited disease. New York:
McGraw-Hill, 1995: 2785-2823.
-
Okhura T, Yamashita K, Kobata A. Urinary oligosaccharides of
GM1-gangliosidosis. J Biol Chem 1981; 256: 8485-8490.
-
Warner ThG, Robertson AD, O’Brien JS. Diagnosis of
GM1-gangliosidosis based on detection of urinary
oligosaccharides with high performance liquid chromatography.
Clin Chem Acta 1983; 127: 313-326.
-
Sandhoff K, Andreae U, Jatzkewitz H. Deficient hexosaminidase
activity in an exceptional case of Tay-Sachs disease with
additional storage of kidney globoside in visceral
organs. Life Sci 1968; 7: 283-288.
-
Gravel RA, Clarke JTR, Kaback MM, Mahuran D, SandhoffK, Suzuki
K. The GM2 gangliosidosis. In: Scriver CR, Beaudet AL, Sly WS,
Valle D, eds. The metabolic and molecular bases of inherited
disease. New York: McGraw-Hill, 1995: 2839-2881.
-
Strecker G, Herlant-Peers MC, Fournet B, Montreuil J, Dorland
L, Haverkamp J, et al. Structure of seven oligosaccharides
excreted in the urine of a patient with Sandhoff’s disease
(GM2-gangliosidosis-variant O). Eur J Biochem 1977; 81:
165-171.
-
Warner ThG, de Kremer RD, Applegarth D, Mock AK. Diagnosis and
characterization of GM2 gangliosidosis type II (Sandhoff
disease) by analysis of accumulating Nacetyl-glucosaminyl
oligosaccharides with high performance liquid chromatography.
Clin Chim Acta 1986; 154:151-164.
-
Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver
CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and
molecular bases of inherited disease. New York: McGraw-Hill,
1995: 2465-2493.
-
Lorincz AE, Hurst RE, Kolodny EH. The early laboratory
diagnosis of mucopolysaccharidoses. Ann Clin Lab Sci 1982; 12:
258-266.
-
De Jong JGN, Wevers RA, Laarakkers C, Poorthuis BJHM.
Dimethylmethylene blue based spectrophotometry of
glycosaminoglycans in untreated urine: a rapid screening
procedure for mucopolysaccharidosis. Clin Che 1989; 35:
1472-1477.
-
De Jong JG, Wevers RA, Liebrand-van Sambeek R. Measuring
urinary glycosaminoglycans in the presence of protein: an
improved screening procedure for mucopolysaccharidoses based
on dimethylmethylene blue. Clin Chem 1992; 38: 803-807.
-
Hopwood JJ, Harrison JR. High-resolution electrophoresis of
urinary glycosaminoglcyans: an improved screening test for the
mucopolysaccharidoses. Anal Biochem 1982; 110: 120-127.
-
Ullrich K, Kresse H. Mucopolysaccharidoses. In: Blau N, Duran
M, Blaskovics M, eds. Physician’s guide to the laboratory
diagnosis of metabolic diseases. London: Chapman & Hall,
1996: 303-319.
-
Barth ML, Ward C, Harris A, Saad A, Fensom A. Frequency of
arylsulphatase A pseudodeficiency associated mutations in a
healthy population. J Med Genet 1994; 31: 667-671.
-
Guo Y, He W, Boer AM, Wevers RA, de Bruijn AM, Groener JE,
Hollak CE, Aerts JM, Galjaard H, van Diggelen OP. Elevated
plasma chitotriosidase activity in various lysosomal storage
disorders. J Inher Metab Dis 1995; 18: 717-722.
Laboratorium Kindergeneeskunde en Neurologie, Academisch
Ziekenhuis Nijmegen, Postbus 9101, 6500 HB Nijmegen
Correspondentie: Dr. J.G.N. de Jong, Academisch Ziekenhuis
Nijmegen, 319 Laboratorium voor Kindergeneeskunde en
Neurologie, Reinier Postlaan 4, 6525 GC Nijmegen.
E-mail: j.dejong@ckslkn.azn.nl

|
Hoofdmenu |
|