|
Ned Tijdschr Klin Chem 2000; 25: 27-30
L.P. van den HEUVEL, J.M.F. TRIJBELS, A.J.M. JANSSEN en J.A.M.
SMEITINK
Mitochondriële encephalomyopathieën zijn ziekten veroorzaakt door
structurele en/of functionele afwijkingen van de mitochondriën.
Classificatie vindt onder andere plaats op basis van het
biochemisch defect.
Er bestaat een grote variabiliteit in klinische expressie en
structurele en functionele mitochondriële afwijkingen. Een
nauwkeurige diagnose is noodzakelijk voor het verstrekken van
behandelingsen erfelijkheidsadviezen.
De waarde van de bepaling van lactaat en van andere metabolieten
voor de diagnostiek van mitochondriële encephalomyopathieën wordt
besproken.
Mitochondriën spelen als productie-eenheden van energie een zeer
belangrijke rol bij het functioneren van cellen. De energie is
nodig voor een uitgebreid scala van processen, zoals
warmteproductie, synthese- en transportprocessen. In de
spiercellen vereist de contractie veel energie. Deze energie wordt
in de vorm van ATP via twee routes in de spiercel aangemaakt: via
de glycolyse in het cytoplasma van de cel en via de oxydatieve
fosforylering in de mitochondriën.
Gedurende de laatste decennia zijn veel patiënten beschreven met
een stoornis in de mitochondriële energievoorziening van de cel
(1,2). Deze defecten werden met name aangetoond bij patiënten
lijdend aan een zogenaamde mitochondriële myopathie, een ziekte
welke gedefinieerd wordt als spierziekte, gekarakteriseerd door
structureel afwijkende mitochondriën en/of abnormaal
functionerende mitochondriën.
Daar mitochondriën in alle lichaamscellen behalve erytrocyten
voorkomen kan in principe ieder orgaan of weefsel zijn aangedaan.
Frequent voorkomende klinische symptomen zijn: spierzwakte,
inspanningsintolerantie, hypotonie, cardiomyopathie en
aandoeningen van het centrale zenuwstelsel. Uit deze opsomming kan
worden afgeleid dat de spierziekte geïsoleerd maar ook als deel
van een "multisystem disorder" kan voorkomen.
Biochemisch worden mitochondriële myopathieën gekarakteriseerd
door stoornissen in de mitochondriële energievoorziening. Deze
stoornissen kunnen worden onderverdeeld in:
-
Defecten in het transport over het mitochondriële membraan. Als
voorbeeld zij genoemd deficiëntie van het carnitine
transportsysteem, waardoor langketen vetzuren
intramitochondriëel niet geoxideerd kunnen worden, alsmede
deficiënties van de adenine nucleotide translocator (ANT) en
"voltage dependent anion channel" (VDAC) (3).
-
Defecten in de mitochondriële substraatoxidatie. Als voorbeeld
kan genoemd worden deficiëntie van één van de enzymen van het
pyruvaat-dehydrogenase- complex, waardoor de oxidatie van
pyruvaat gestoord is.
-
Defecten in één of meer complexen van de mitochondriële
ademhalingsketen. Inmiddels zijn defecten in alle complexen
beschreven.
-
Defecten gepaard gaande met een stoornis in de
energieconservering. Het meest bekende voorbeeld in deze
categorie is de "Luft disease", waarbij sprake is van
losse koppeling van oxidatie en fosforylering (4).
De oxidatieve fosforylering:
Mitochondriën zijn subcellulaire organellen die voorkomen in alle
kernhoudende cellen. Ze bestaan uit een glad buitenmembraan, een
intermembraan ruimte, een geplooid binnenmembraan en een matrix.
Het buitenmembraan heeft poriën waardoor kleine moleculen vrij
kunnen diffunderen. Het binnenmembraan is ondoorlaatbaar voor de
meeste moleculen. Het bevat translocatoren voor transport van
onder andere ionen en eiwitten.
Een belangrijke functie van mitochondriën is de oxidatieve
fosforylering (OXFOS). Hieronder wordt verstaan de oxidatie van
brandstoffen en de productie van energie in de vorm van
adenosine-triphosphaat (ATP).
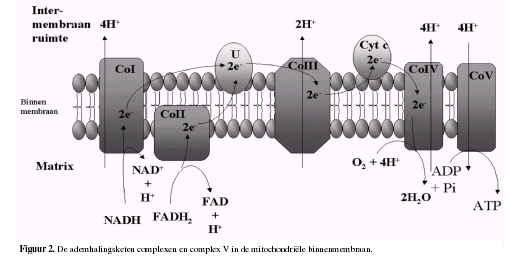
De elektronentransportketen (complex I-IV, coenzym Q en cytochroom
c) en het ATP-synthase (complex V), gelegen in de dubbele
lipidelaag van de mitochondriële binnenmembraan, vormen
gezamenlijk het OXFOS-systeem (Figuur 1).
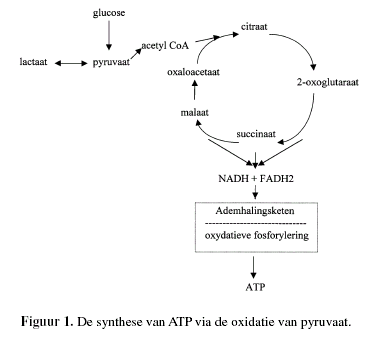
Het via de citroenzuurcyclus gevormde NADH en FADH2 wordt
gedehydrogeneerd via respectievelijkcomplex I en II van de
elektronentransportketen tot NAD+ en FAD.
De hierbij vrijkomende elektronen worden via de diverse
elektronentransportketen complexen en de beide elektronencarriers
(coenzym Q en cytochroom c) getransporteerd en reduceren
uiteindelijk zuurstof tot water.
Dit elektronentransport bevordert tevens de uitstoot van protonen
uit de matrix door het binnenmembraan naar de intermembraanruimte.
Deze protonen keren terug via complex V, bestaande uit een
protonenkanaal en het ATP-synthase, het enzym dat de synthese van
ATP uit adenosine-difosfaat (ADP) en anorganisch fosfaat (Pi)
katalyseert(Figuur 2).
Metabool onderzoek van lichaamsvloeistoffen:
De leeftijd waarop de eerste symptomen van OXFOS defecten
duidelijk worden en het beloop van deze aandoeningen is
buitengewoon heterogeen. Dit wordt ten dele verklaard door de
verschillen in de genetische oorzaak. Daar mitochondriën in alle
kernhoudende cellen voorkomen kan in principe ieder weefsel of
orgaan zijn aangedaan. De meest frequent aangedane weefsels en
organen zijn die welke een hoge energie-behoefte hebben zoals
hersenen, hartspier en skeletspier. Vandaar dat deze groep van
ziekten ook wel als mitochondriële encephalomyopathieën omschreven
wordt.
Bij verdenking op mitochondriële (encephalo)myopathie moet in
ieder geval gestart worden met uitgebreid klinisch-chemisch
onderzoek van lichaamsvloeistoffen alvorens weefsel wordt
onderzocht.
De meest relevante parameter hierbij vormt lactaat. Bij een
storing in het OXFOS neemt zowel de cytosolische NADH/NAD+ - als
mitochondriële NADH/NAD+ ratio toe. Aangezien het pyruvaat
dehydrogenase complex feed-back remming ondervindt onder invloed
van een verhoogde NADH/ NAD+ ratio, zal de concentratie van
pyruvaat dientengevolge toenemen en tevens van lactaat dat in het
cytosol uit pyruvaat wordt gevormd via lactaat dehydrogenase.
Tevens kan de cytosolische lactaat/pyruvaat ratio toenemen. De
theoretische verklaring kan uit onderstaande vergelijking worden
afgeleid. De evenwichtsconstante (K) voor de omzetting van
pyruvaat naar lactaat is: K = [Lactaat]
[NAD+]/[Pyruvaat][NADH][H+] Daar de evenwichtsconstante (K) van
deze reactie constant blijft, kan bij een stoornis in de OXFOS,
waarbij de NADH/NAD+ ratio verhoogd is, de evenwichtsligging in de
richting van lactaat verschoven worden. Hierdoor ontstaat een
verhoogde ratio van lactaat/pyruvaat.
De lactaat/pyruvaat ratio is met name verhoogd bij ernstige
stoornissen in één of meer complexen van de mitochondriële
ademhalingsketen. Bij een deficiëntie van het pyruvaat
dehydrogenase complex wordt een normale lactaat/pyruvaat ratio
gevormd (normaalwaarde < 20).
Bepaling van de veelal gepropageerde ratio 3-hydroxybutyraat/
acetoacetaat heeft geen toegevoegde waarde in de
klinisch-chemische vóórscreening naast de lactaat/pyruvaat
ratio.
Indien de lactaatconcentratie in plasma de nierdrempel (5-7 mM)
overschrijdt, zal de lactaatconcentratie in de urine eveneens
toenemen.
Biochemische screening procedures omvatten analysen van de
volgende metabolieten in lichaamsvloeistoffen: L-lactaat (bloed,
urine en eventueel liquor), aminozuren (serum, urine), vrij en
totaal carnitine (bloed) en organische zuren (urine).
Indien het centrale zenuwstelsel klinisch betrokken is bij de
pathologie, moet ook het lactaatgehalte in de liquor geanalyseerd
worden.
L-lactaat is de meest informatieve metaboliet om te testen. Het
kan verhoogd zijn in lichaamsvloeistoffen van patiënten met
pyruvaat dehydrogenase complex deficiëntie of een defect in de
oxidatieve fosforylering.
n welke lichaamsvloeistof moet lactaat bepaald worden? Bloed
lactaatgehalten kunnen sterk variëren bij een patiënt en
reflecteren vaak de ernst van het ziektebeeld. Het lactaatgehalte
in het bloed wordt beïnvloed door fysieke inspanning, voeding en
stress. Het verdient de voorkeur om in het bloed lactaatgehalten
meerdere malen vóór en na de voeding te analyseren.
Ervaring van de eigen groep heeft aangetoond dat verzet tegen een
vena punctie, met name bij kinderen, kan resulteren in een
vijfvoudig verhoogd lactaatgehalte in het bloed. Om betrouwbare
lactaatgehalten in het bloed te meten bij kinderen wordt
geadviseerd een intraveneuze cannula in te brengen. Na 45 minuten
bedrust kan het bloedmonster voor analyse afgenomen worden.
Het verdient aanbeveling de bepaling van lactaat zowel in bloed
als urine (verzameld tussen 08.00 en 17.00 uur) te verrichten.
Wanneer echte lactaatacidurie waargenomen wordt, moet ook lactaat
in het bloed bepaald worden aangezien verhoogde excretie van
lactaat in de urine ook veroorzaakt kan worden door een
verminderde tubulaire reabsorptie.
Patiënten (waarbij uiteindelijk een mitochondriopathie is
vastgesteld) met een lactaatacidurie en normale lactaatgehalten in
het bloed zijn bekend.
Ook de omgekeerde situatie (dat wil zeggen normaal lactaatgehalte
in urine ondanks lactaatacidemie) is beschreven.
Zelfs patiënten met noch lactaatverhoging in het bloed noch in de
urine zijn bekend. Deze patiënten zijn dan gebiopteerd op
klinische gronden alleen, hetgeen eens te meer duidt op het belang
van een integraal onderzoek.
De ervaring van de eigen groep bij patiënten met een enzymatisch
bewezen defect in het pyruvaat dehydrogenase complex en/of in de
complexen van de mitochondriële ademhalingsketen is als volgt: bij
25% van deze patiënten werd alleen het lactaatgehalte in het bloed
gemeten. In 85% van deze patiënten was lactaat verhoogd. In 75%
van de patiënten werd zowel het lactaatgehalte in het bloed als in
de urine gemeten: 40% van deze patiënten vertoonden verhoogde
gehalten in zowel bloed als urine, 10% alleen in het bloed, 20%
van de patiënten alleen in de urine en 30% van de patiënten
vertoonde noch in het bloed noch in de urine verhoogde
lactaatgehalten.
Bij sommige patiënten met verhoogd lactaatgehalte in het bloed
werden waarden waargenomen variërend van normaal tot duidelijk
verhoogd, hetgeen eens te meer de noodzaak toont van herhaalde
analysen. Lactaatgehalten in urine zijn leeftijdsafhankelijk
(tabel 1) (5).

Lamers et al. (6) vonden echter geen leeftijdsafhankelijk effect
voor bloed lactaatgehalten bij twee leeftijdsgroepen (3-5 jaar
en 6-15 jaar) na 14 uur vasten.
De eerder genoemde data tonen duidelijk aan dat bepaling van
bloed en/of urine lactaatgehalten geen volledig betrouwbare
screeningsprocedure vormt. Echter de liquor lactaatconcentratie
bij de bovenstaande patiëntengroep bleek bij meer dan 90% van de
patiënten verhoogd te zijn. Dit betekent dat de lactaatbepaling
in liquor de meest betrouwbare diagnostische procedure vormt,
met name bij patiënten met neurologische symptomen.
Gegevens over leeftijdsafhankelijkheid van lactaatgehalten in
liquor zijn momenteel niet voorhanden. Veelal wordt gelijktijdig
lactaat en pyruvaat gemeten, aangezien defecten in OXFOS vaak
geassociëerd zijn met verhoogde lactaat/ pyruvaat ratio in bloed
en CSF.
Echter onze ervaring is dat een normale lactaat/pyruvaat ratio
een stoornis in de electronentransportketen niet uitsluit.
Bepaling van alanine in bloed en liquor is eveneens aan te
bevelen bij de screening van patiënten met een
mitochondriopathie aangezien hyperalaninemie kan duiden op
lactaatacidemie. Deze toename in het alaninegehalte in het bloed
wordt veroorzaakt door transaminering van geaccumuleerd pyruvaat
naar alanine. Echter patiënten met duidelijk verhoogd lactaat in
het bloed kunnen normale alanine concentraties in het bloed
vertonen.
Bepaling van aminozuren en organische zuren in urine van
verdachte patiënten is eveneens zinvol, aangezien soms een
gegeneraliseerde aminoacidurie en/of verhoogde excretie van
organische zuren wordt vastgesteld.
Ten aanzien van de metabole vóórscreening van verdachte
patiënten kan derhalve gesteld worden dat bepaling van lactaat,
pyruvaat en alanine in bloed en liquor (dit laatste uiteraard
alleen bij patiënten met een encephalopathie) en van lactaat,
aminozuren en organische zuren in urine een goede basis vormt
voor selectie van patiënten waarbij nadere biochemische
diagnostiek geïndiceerd is. Deze biochemische diagnostiek
bestaat uit meting van de oxidatieve capaciteit van de
mitochondriële ademhalingsketen en bepaling van de activiteit
van enzymen van de ademhalingsketen.
In vivo functie testen:
Sommige patiënten, die op grond van klinische symptomen verdacht
worden van een mitochondriële encephalomyopathie, vertonen geen of
slechts geringe lactaatacidemie en/of lactaatacidurie.
Lactaatconcentraties in liquor zijn eveneens niet altijd duidelijk
afwijkend. In deze patiënten kan een
in vivo functie test (met name de glucose tolerantie test)
uitkomst bieden.
Het gebruik van de pyruvaat belastingstest (7) wordt ontraden
gezien een bijna fatale afloop van deze diagnostische test in de
eigen kliniek. De orale glucose tolerantie test wordt uitgevoerd
volgens een vast protocol.
Normaal wordt geen of slechts een geringe stijging van het bloed
lactaatgehalte waargenomen na belasting, terwijl bij patiënten met
een defect in de pyruvaat oxidatie duidelijk een toename (tweemaal
de uitgangswaarde!) wordt waargenomen.
Het dient echter benadrukt te worden dat een normale glucose
tolerantie test een mitochondrieel defect niet geheel uitsluit.
Tenslotte:
Vele valkuilen kunnen leiden tot een vals-positieve of
vals-negatieve diagnose bij mitochondriële myopathieën. Vaak moeten
klinische, biochemische en morfologische aspecten in combinatie
beoordeeld worden om diagnostische fouten te voorkomen.
Lactaat aanwezig in lichaamsvloeistoffen is de meest informatieve
metaboliet om te testen bij verdenking op een mitochondriële
myopathie. Bij voorkeur vindt de lactaatbepaling gelijktijdig plaats
in bloed, urine en liquor.
In bloed en liquor wordt tevens pyruvaat bepaald, aangezien een
verhoogde lactaat/pyruvaat ratio een extra indicatie vormt voor een
defect in de ademhalingsketen. De lactaatbepaling in de liquor vormt
de meest betrouwbare diagnostische procedure. Normale
lactaatgehalten sluiten een mitochondriële myopathie echter niet
uit.
Onderzoek noodzakelijk voor een juiste diagnose is complex en dient
bij voorkeur plaats te vinden in een centrum met de benodigde
expertise. Op basis van de klinische gegevens en van het
klinisch-chemisch vóóronderzoek vindt aansluitend geavanceerde
biochemische en moleculair genetische diagnostiek plaats, bij
voorkeur in spierweefsel, om een eventueel defect op enzym-
en/of DNA niveau te localiseren.
Literatuur:
1. Shoffner JM. Maternal inheritance and the evaluation of
oxidative phosphorylation diseases. Lancet 1996; 348:
1283-1288.
2. Smeitink J, Van den Heuvel L. Human complex I in health and
disease. Am J Hum Genet 1999; 64: 1501-1510.
3. Huizing M, Ruitenbeek W, Thinnes FP, DePinto V, Wendel U,
Trijbels FJM, Smit LME, Ter Laak HJ, Van den Heuvel LP. Deficiency
of the Voltage-Dependent Anion Channel: A novel cause of
mitochondriocytopathy. Pediatr Res 1996;39: 760-765.
4. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B. A case of
severe hypermetabolism of nonthyroid origin with a defect in the
maintenance of mitochondrial respiratory control: correlated
clinical, biochemical, and morphological study. J Clin Invest
1962; 41: 1776-1780.
5. Trijbels JMF, Scholte HR, Ruitenbeek W, Sengers RCA, Janssen
AJM, Busch HFM. Problems with the biochemical diagnosis in
mitochondrial (encephalo-)myopathies. Eur J Pediatr 1993; 152:
178-184.
6. Lamers KJB, Doesburg WH, Gabreëls FJM, Lemmens WAJG, Romsom AC,
Wevers RA, Reinier WO. The concentration of blood components
related to fuel metabolism during prolonged fasting in children.
Clin Chim Acta 1985; 152: 155-163.
7. Erven PMM, Gabreëls FJM, Wevers RA, Doesburg WH, Ruitenbeek W,
Reinier WO, Lamers KJB. Intravenous pyruvate loading test in Leigh
syndrome. J Neurol Sci 1987; 77: 217-227.
Laboratorium voor Kindergeneeskunde & Neurologie, Centrum voor
Mitochondriële Ziekten, Academisch Ziekenhuis Nijmegen
Correspondentie: Dr. L.P. van den Heuvel, Laboratorium
Kindergeneeskunde & Neurologie (424), Academisch Ziekenhuis
Nijmegen, Postbus 9101, 6500 HB Nijmegen.

|
Hoofdmenu |
|
