Peroxisomen
|
|
|
Peroxisomen
|
|
|
|

|
Elektron Microscopic Atlas : Peroxisomen |
De peroxisomale bèta-oxidatie verloopt van enoyl-CoA via D-3-hydroxyacyl-CoA tot 3-ketoacyl-CoA door de actie van de D-3-hydroxyacyl-CoA dehydratase/D-3-hydroxyacyl-CoA dehydrogenase "bifunctional protein"; ook wel D-bifunctional protein ( OMIM: 601860 ) genoemd.
3-methyl vertakte vetzuren zoals phytaanzuur kunnen niet in de
β-oxidatie worden afgebroken, omdat de 3-methyl groep
de dehydrogenering blokkeert van de hydroxylgroep door
hydroxyacyl-CoA dehydrogenase. De oplossing voor dit dilemma is
de ά-oxidatie.
Alfa-oxidatie is het proces waarbij vetzuren door een oxidatieve
decarboxylatie met 1 koolstofatoom verkort worden.
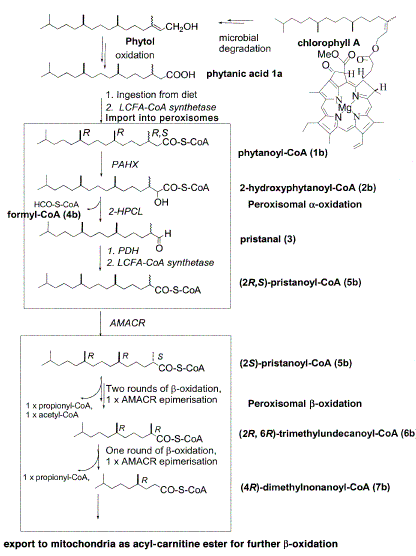
De afbraak van phytaan- en pristaanzuren
in peroxisomen door de
ά- en β oxidatie routes
LCFA-CoAsynthetase,
long-chain fatty acyl-CoA synthetase;
PAHX, phytanoyl-CoA 2-hydroxylase;
2HPCL,
2-hydroxyphytanoyl-CoA lyase;
PDH, pristanal dehydrogenase;
AMACR, a-methylacyl-CoA racemase.
| ExPASy | |
| acyl-CoA oxidase | EC 1.3.3.6 |
| D-bifunctional protein | OMIM 601860 |
| 3-ketoacyl-CoA thiolase | EC 2.3.1.16 |
| D3,5-D2,4-dienoyl-CoA isomerase | |
| catalase | EC 1.11.1.6 |
| serine-pyruvate aminotransferase | EC 2.6.1.51 |
| carnitine-o-acetyltransferase | EC 2.3.1.7 |
| D-aspartate oxidase | EC 1.4.3.1 |
| phosphomevalonate kinase | EC 2.7.4.2 |
| thioesterase | |
| mevalonate kinase | EC 2.7.1.36 |
| dihydroxyacetonephosphate acyltransferase | EC 2.3.1.42 |
| Microsomal epoxide hydrolase | EC 3.3.2.9 |
| phytanoyl-CoA alpha-hydroxylase | EC 1.14.11.18 |
| isocitrate dehydrogenase | EC 1.1.1.42 |
| D3,D2-enoyl-CoA isomerase | EC 5.3.3.8 |
| malonyl-CoA decarboxylase | EC 4.1.1.9 |
| sterol-carrier protein 2 |
Daar peroxisomen geen DNA of ribosomen hebben, moeten al hun
proteïnen geïmporteerd worden.
Omdat de levensduur van een peroxisoom ongeveer 2 dagen is, vindt
er een continu proces van vorming en vervanging plaats. Dit proces
van vorming en vervanging wordt de " peroxisomale biogenese
" genoemd.
Lazarow en Fujiki toonden aan dat nieuwe peroxisomale
proteïnen worden gesynthetiseerd op vrije polyribosomen en
concludeerden dat peroxisomen samen met mitochondriën en chloroplasten tot een groep van autonome organellen hoorden
die zich vermenigvuldigen door groei en deling (1985).
Echter, dankzij moderne electronenmicroscopische technieken en
real time imaging is heel mooi te zien dat peroxisomen zonder
enige twijfel ontstaan vanuit het ER. ( endoplasmatisch reticulum
).
De geboorte van peroxisomen is bijzonder goed zichtbaar in
dendritische (afweer)cellen van de muis.
Immuno-electronmicroscopie toonde aan dat een bepaald type
peroxisomsal membraaneiwit (Pex13) juist in deze cellen wordt
gevonden op gespecialiseerde gebieden van het ER, in lamel-achtige
structuren en in iets mindere mate in volgroeide peroxisomen zelf.
Typerende peroxisomale enzymen worden daarentegen alleen in de
eicel-vormige volgroeide peroxisomen gevonden. In
3D-reconstructies dankzij electronen tomograpie kon de
membraancontinuïteit tussen gespecialiseerd ER, lamellen en
volgroeide peroxisomen worden aangetoond.
De assemblage van het peroxisoom:
Het peroxisoom is een compartiment omgeven door een membraan. Dit membraan bestaat uit meerdere proteïnen die essentieel zijn voor de functie van het peroxisoom. De ruimte binnen het membraan wordt de peroxisomale matrix genoemd. Ook de matrix bevat vele noodzakelijke proteïnen voor de goede functie van het peroxisoom.
De vorming van het peroxisoom bestaat uit een aantal stappen:
De eerste stap is de vorming van het peroxisomale membraan en de
import van membraanproteïnen in het membraan. Deze proteïnen
worden buiten het peroxisoom gemaakt, vervolgens naar het
peroxisoom getransporteerd en dan in het membraan geplaatst.
De laatste stap is de import van de peroxisomale matrixproteïnen
over de peroxisomale membraan in de peroxisomale matrix.
Om het bovenstaande proces normaal te laten verlopen zijn er
op zijn minst 12 menselijke proteïnen nodig. Deze proteïnen
worden peroxins worden genoemd. (Distel et al.,
1996). Deze peroxins worden PEX1, PEX2, PEX3 etc. genoemd.
Een defect in een van deze 12 peroxins resulteert in een niet
functionerend peroxisoom en leidt tot een groep ziekten die bekend
staat als de Peroxisomale Biogenese Ziekten.
Proteïnen die bestemd zijn voor het peroxisoom bevatten een
signaal: de eerste is een peroxisomal signal type 1 ( PTS 1); of
PTS2.
Men denkt dat er drie proteïnen betrokken zijn bij de
vorming van het peroxisomale membraan ( PEX3, PEX 16 en PEX19).
Een defect in een van deze drie proteïnen zal er toe leiden dat er
geen membraan gevormd kan worden, waarbij er dan ook geen
proteïnen geïmporteerd kunnen worden die het PTS1 of PTS2 signaal
bevatten.
Peroxisomale matrixproteïnen worden buiten het peroxisoom gemaakt
en bevatten of het PTS1 signaal, of het PTS2 signaal. Deze
proteïnen worden herkend door receptoren; dit zijn proteïnen die
verantwoordelijk zijn voor het vervoer van proteïnen met een PTS
signaal naar het peroxisoom.
Deze receptoren worden PEX5 genoemd ( herkent PTS1) en PEX7
(herkent PTS2).
Deze receptoren vervoeren de proteïnen naar het peroxisomale
membraan. Vervolgens geven de receptoren via een aantal stappen (
en over het membraan ) de proteïnen af aan de matrix.
Het grote belang van peroxisomen in het metabolisme van een gezonde cel wordt waarschijnlijk het best geïllustreerd door het voorkomen van enkele zeer ernstige, soms zelfs dodelijke, aandoeningen bij de mens die worden veroorzaakt door afwijkingen in het functioneren van peroxisomen. De verzamelnaam voor deze aandoeningen is PBD (Peroxisome Biogenesis Disorders).
Gisten zijn zeer geschikte organismen voor het bestuderen van de vorming (biogenese) van peroxisomen. Mutanten van deze organismen, waarin dit proces verstoord is (zogenaamde pex mutanten) zijn namelijk levensvatbaar, mits gekweekt onder de juiste condities. Dit maakt het mogelijk om het defecte gen en het bijbehorende eiwitproduct te identificeren. Op deze wijze zijn al 25 eiwitten die direct betrokken zijn bij peroxysoom biogenese, de zogenaamde “peroxins”, ontdekt.
Gisten zijn bij uitstek geschikt als modelorganismen voor de studie naar de oorzaken van PBD’s. Op basis van informatie uit het onderzoek naar peroxysoom biogenese in gisten, zijn van 11 van de 12 nu bekende PBD’s de oorzaken op moleculair niveau bekend.
Bij onderzoek naar de biogenese van peroxisomen is jarenlang
uitgegaan van het “growth and fission” (“groei en deling”) model
als verklaring voor de wijze van vermeerdering van deze
organellen. Dit model stelt, dat nieuwe peroxisomen worden
gevormd uit reeds bestaande organellen, door een proces van
groei gevolgd door deling. In geval van zich delende cellen zijn
de peroxisomen in de dochtercel via deling elk afkomstig van de
moedercel. Dit model sluit de vorming van nieuwe peroxisomen
de novo (“uit het niets”) uit.
Diverse recente onderzoeksresultaten geven aanleiding om aan het
groei en deling model te twijfelen. Deze resultaten lijken erop
te wijzen dat in ieder geval in een aantal organismen de
mogelijkheid van een alternatieve herkomst van peroxisomen niet
kan worden uitgesloten.
Bij de mens is een aantal ernstige, genetische ziekten bekend
waarbij er iets mis is met het peroxisoom. Zo hebben patiënten met
het Zellwegersyndroom geen peroxisomen in hun cellen.
Bij een andere peroxisomale afwijking, RCDP (rhizomelic
chondrodysplasia punctata), zijn deze compartimenten wel aanwezig,
maar functioneren ze niet volledig.
Kinderen die met dit soort zeldzame stofwisselingsziekten worden
geboren hebben een grote ontwikkelingsachterstand en leven vaak
minder dan een jaar. De patiënten blijken zeer weinig etherlipiden
in hun lichaam te hebben. Hoewel de relatie tussen de symptomen en
het ontbreken van etherlipiden niet duidelijk is, kan het
detecteren van de lipiden wel een hulpmiddel zijn bij het
vaststellen van deze ziekten.
Peroxisomale Biogenese Ziekten zijn ziekten waarbij het proces van
de nieuwe vorming van peroxisomen slecht functioneert en waarbij
bijna alle normale peroxisomale functies afwezig - of deficiënt
zijn.
Soms houdt dit in dat het peroxisoom niet gevormd kan worden; soms
houdt dit in dat de peroxisomen niet in voldoende hoeveelheden
gevormd kan worden ; soms houdt dit in dat ze wel gevormd worden ,
maar de enzymen missen die nodig zijn om te functioneren.
Peroxisomale biogenese ziekten zijn o.a. Zellweger Syndrome ,
Neonatal Adrenoleukodystrophy en Infantile Refsum Disease.
Peroxisomale Multi-Enzym Ziekten zijn ziekten waarbij meerdere
enzymen in het peroxisoom deficiënt zijn zonder dat er sprake is
van een algeheel verlies van de functie van het peroxisoom , zoals
bij de peroxisomale biogenese ziekten.
Peroxisomale multi-enzym ziekten zijn o.a Rhizomelic
chondrodysplasia punctata en Zellweger-Like Syndrome.
Peroxisomale Single-Enzyme Ziekten zijn ziekten waarbij het
peroxisoom intact is en functioneert, maar waarbij er een defect
is in één enzymatisch proces.
Peroxisomale Single-Enzyme Ziekten zijn o.a. X-ALD, Peroxisomal
Thiolase Deficiency, Acyl CoA Oxidase Deficiency, Bifunctional
Protein Deficiency, DHAP-AT Deficiency, Alkyl DHAP Synthase
Deficiency, Glutaryl CoA Oxidase Deficiency , Mevalonate Kinase
Deficiency, Hyperoxaluria Type I , Acatalasemia en classical or
adult onset Refsum Disease.
Extra informatie:
Peroxisomal fatty acid alpha- and beta oxidation in
humans
Zellweger Syndrome ( Ziekte van Zellweger
)
Synoniemen: ZS; Cerebrohepatorenal Syndrome;
CHR Syndrome; ZWS.
Who Named It?: Hans Ulrich Zellweger
OMIM:
214100
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Pseudo Zellweger Syndrome ( Peroxisomal
3-oxoacyl CoA Thiolase Deficiency )
Deze stoornis is nu ondergebracht onder: D-Bifunctional
protein Deficiency
D-Bifunctional protein deficiency
Synoniemen voor deze stoornis zijn: Deficiency of
17-abeta-hydroxysteroid dehydrogenase IV; DBP deficiency;
Peroxisomal bifunctional enzyme deficiency; PBFE
deficiency.
OMIM:
261515
OMIM
Clinical Synopsis
OMIM:
17-abeta-hydroxysteroid dehydrogenase IV
e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Adrenoleukodystrophy, autosomal neonatal form ( neonatale adrenoleukodystrofie )
Synoniemen voor deze ziekte zijn: Neonatal
adrenoleukodystrophy; NALD.
OMIM:
202370
OMIM:
Clinical Synopsis
e-medicine:
Peroxisomal disorders
The peroxisome website
Adrenoleukodystrophy; ALD ( X-chromosoom gebonden adrenoleukodystrofie )
Synoniemen voor deze ziekte zijn: Addison disease and cerebral
sclerosis; Adrenomyeloneuropathy; AMN;
Siemerling-Creutzfeldt disease; Bronze Schilder disease;
Melanodermic leukodystrophy.
OMIM:
300100
OMIM:
Clinical Synopsis
Extra informatie:
Belangenvereniging X-ALD
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Pseudoneonatal Adrenoleukodystrophy
Synoniemen voor deze ziekte zijn: Peroxisomal Acyl CoA Oxidase
deficiency; Pseudo neonatal adrenoleukodystrophy.
OMIM:
264470
ExPASy: Acyl-CoA oxidase.
EC 1.3.3.6
Refsum disease, infantile form
(
Infantiele Refsum ziekte of infantiele phytaanzuur
stapelingsziekte )
Synoniemen voor deze ziekte zijn: IRD; Infantile
Phytanic acid storage disease.
OMIM:
266510
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Extra informatie:
Refsum's disease: a peroxisomal disorder affecting phytanic
acid a-oxidation
Dit is een pdf-document
Refsum disease ( Ziekte van Refsum of
phytaanzuur oxidase deficiëntie )
Synoniemen voor deze ziekte zijn; Phytanic acid oxidase
deficiency; Heredopathia Atactica Polyneuritiformis;
Hereditary motor and sensory neuropathy IV; HMSN IV.
OMIM:
266500
OMIM:
Clinical Synopsis
OMIM:
phytanoyl-CoA hydroxylase
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Extra informatie:
Refsum's disease: a peroxisomal disorder affecting phytanic
acid a-oxidation
Dit is een pdf-document
Rhizomelische Chondrodysplasia Punctata,
Type I; RCDP I
Synoniemen voor deze ziekte zijn: Chondrodysplasia Punctata,
Rhizomelic Form; CDPR; Chondrodystrophia
CalcificansPunctata.
OMIM:
215100
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie: The peroxisome website
Extra informatie:
Nederlandse RCDP pagina
Glutaryl CoA-Oxidase Deficiency
Synoniemen voor deze ziekte zijn: Glutaricaciduria III; GA
III
OMIM:
231690
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
Hyperoxaluria, Primary, Type II (
Primaire hyperoxaluria type II )
Synoniemen voor deze ziekte zijn: HP2; Oxalosis II;
Glycericaciduria; glyoxylate reductase/hydroxypyruvate
reductase deficiency;D-Glycerate Dehydrogenase deficiency.
OMIM:
260000
OMIM:
Clinical Synopsis
OMIM:
glyoxylate reductase/hydroxypyruvate reductase
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website
ExPASy: Glyoxylate reductase (NADP+).
EC 1.1.1.79
Glycolate + NADP+ <=>
glyoxylate + NADPH
Mevalonicaciduria (
Mevalon acidurie )
OMIM:
610377
OMIM Mevalonate Kinase:
251170
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie: The peroxisome website
ExPASy: Mevalonate Kinase
EC 2.7.1.3
ATP + (R)-mevalonate <=> ADP +
(R)-5-phosphomevalonate
Hyperpipecolatemia (
hyperpipecoline acidemie )
Synoniemen voor deze ziekte zijn: Hyperpipecolicacidemia
OMIM:
239400
OMIM:
Clinical Synopsis
Extra informatie: e-medicine:
Peroxisomal disorders
Extra informatie:
The peroxisome website

|
Hoofdmenu |
|
|
|
|
|
|

