Lysosomen:
Lusis ( Grieks ) betekent losmaken en
sooma betekent lichaam.
Lysosomen zijn voor het eerst door de Belgische
biochemicus Christian René de Duve en zijn medewerkers geïsoleerd uit
levercellen van de rat en door hem in 1955 beschreven.
(Sedert 1989) burggraaf ( geboren in Thames
Ditton, Groot Brittanië, 2 oktober 1917), Belgisch biochemicus, werd in
1951 hoogleraar in de fysiologische chemie te Leuven en in 1962 tevens
hoogleraar in de biochemische cytologie aan de Rockefeller University (
New York ).
Hij isoleerde, tezamen met zijn medewerkers, als
eerste (ca 1949/1950) lysosomen ; ook konden de Duve c.s. voor het eerst (
ca 1954/1955 ) met biochemische methoden het peroxisoom identificeren.
In 1974 werd hem, met de biologen A.Claude (Belgie) (
autobiografie ) en G.Palade (V.S.)
(autobiografie
), de Nobelprijs voor fysiologie of geneeskunde toegekend “ voor hun
ontdekkingen betreffende de structurele en functionele organisatie in de
cel “.In 1975 stichtte hij te
Ottignies-Louvain-La-Neuve het
International Institute of Cellular and Molecular
Pathology (ICP).
Lysosomen:
Lysosomen zijn membraan gebonden organellen in
het vloeibare gedeelte (cytoplasma) van de cel , die in alle eukaryote
cellen worden gevonden en nog net zichtbaar zijn onder een
elektronenmicroscoop en die ongeveer 40 verschillende enzymen ( zure
hydrolasen met een pH van ongeveer 5: op een enkele uitzondering na zijn
dit glycoproteïnen die een N-gebonden of soms een O-gebonden
oligosaccharide keten bevatten) waarmee vetten , eiwitten, koolhydraten ,
andere macromoleculen , RNA en DNA worden afgebroken.
Hydrolasen zijn enzymen die hydrolysereacties
katalyseren. Hydrolyse is een splitsing van
scheikundige verbindingen onder opneming van water. Tot deze groep behoren
de peptidasen, esterasen, glycosidasen en de fosfatasen.
De zuur afhankelijke activiteit van de enzymen in
het lysosoom beschermen de cel tegen zelf destructie in het geval het
lysosoom lekt of scheurt, omdat de pH van de cel neutraal tot licht
alkalisch is. ( ongeveer 7.2 ).
Een andere functie van het lysosoom is de afbraak
van bacteriën en organellen die niet meer functioneren, bijvoorbeeld
mitochondriën.
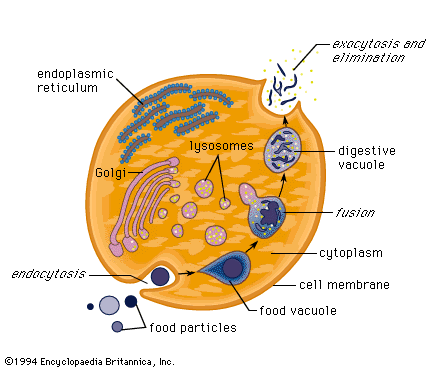
Materialen die in het lysosoom moeten worden
afgebroken , worden hoofdzakelijk op drie manieren naar het lysosoom
getransporteerd : endocytose , autofagocytose en fagocytose.
Bij de endocytose worden extracellulaire
macromoleculen getransporteerd door de celmembraan van buiten de cel naar
binnen , waarbij een membraangebonden vesicula ( zakje ) ontstaat die een
endosoom wordt genoemd en die dan versmelt met een lysosoom.
Bij de endocytose worden verschillende typen
onderscheiden : pinocytose, fagocytose en receptor - mediated endocytose.
Pinocytose is het proces waarbij microscopisch
kleine vloeistofdruppelsdruppels door levende cellen d.m.v. instulpingen (
pinocytoseblaasjes ) van de celmembraan worden opgenomen.
Autofagocytose is het proces waarbij oude
organellen uit de cel worden verwijderd . Oude organellen worden omgeven
door membranen die dan versmelten met een lysosoom.
Fagocytose wordt uitgevoerd door speciale cellen
( macrofagen) die grote extracellulaire deeltjes zoals dode cellen of
vreemde indringers omvatten en dan naar het lysosoom transporteren.
Veel producten van de lysosomale afbraak, zoals
aminozuren en nucleotiden, worden weer teruggebracht in de cel voor de
synthese van nieuwe cellulaire componenten.
Exocytose is het proces waarbij de materialen die
in de cel worden gesynthetiseerd, worden verpakt in membraan gebonden
zakjes en geëxporteerd worden uit de cel , waarbij de zakjes fuseren met
het externe plasma membraan. De geëxporteerde materialen zijn cel
specifieke proteïnen, neurotransmitters en een grote verscheidenheid aan
andere moleculen.
Een cel kan enkele honderden lysosomen bevatten.
Deze lysosomen kunnen verschillen in grootte, vorm en morphologische
verschijning. De grootte varieert van 0,2 tot 2 micrometer in diameter.
De lysosomale enzymen worden gesynthetiseerd door
membraan gebonden ribosomen in het endoplasmatisch retículum. ( Een
netwerk van membranen in het celplasma). Er zijn primaire en
secundaire lysosomen. Primaire lysosomen worden gevormd door het ruwe
endoplasmatisch reticulum en secundaire lysosomen worden gevormd door het
gladde endoplasmatisch reticulum.
Glycosaminoglycanen : ( Mucopolysacchariden )
Muco- betekent amino suiker ; Poly = veel;
sacchariden = koolhydraten. Mucopolysaccharide is een verouderde term voor
glycosaminoglycaan.
De meest voorkomende heteropolysacchariden in het
lichaam zijn de glycosaminoglycanen. Deze moleculen zijn lange onvertakte
polysacchariden die een repeterende disaccharide eenheid ( een aminosuiker
) bevatten. De disaccharide eenheden bevatten of N-acetyl-D-galactosamine
of N-acetyl-D-glucosamine en een uronzuur zoals glucuronzuur of iduronzuur.
Glycosaminoglycanen zijn zeer negatief geladen moleculen.
Glycosaminoglycanen komen voornamelijk voor op de oppervlakte van de
cellen of in de extracellulaire matrix ( ECM ). Glycosaminoglycanen
hebben een hoge viscositeit ( vloeibaarheid ). Glycosaminoglycanen zijn
slecht comprimeerbaar, waardoor deze moleculen uitstekend als
smeervloeistof kunnen dienen in de gewrichten. Door hun stijfheid houden
ze de structuur van de cel in stand en voorzien ze in de mogelijkheid
migratie van cellen toe te staan en de diffusie van water-oplosbare
moleculen.
De meest voorkomende glycosaminoglycaanstructuren
zijn chondroïtine sulfaat , dermatan sulfaat , heparan sulfaat , keratan
sulfaat , hyaluronzuur en heparine.
De meerderheid van de glycosaminoglycanen in het
lichaam zijn verbonden aan een basisproteïne waarbij proteoglycanen worden
gevormd ( ook wel mucopolysacchariden genoemd ). Proteoglycanen en
glycosaminoglycanen vervullen verschillende vitale functies in het
lichaam.
Een bekende functie van het glycosaminoglycaan
heparine is het tegengaan van de bloedstolling. Heparine is een
polysaccharide dat is opgebouwd uit de sulfaatesters van twee
verschillende glucosederivaten. De glucose-eenheden zijn via
α-1,4-bindingen aan elkaar gekoppeld.
Heparine wordt door hogere organismen geproduceerd en komt voor in lever-,
long- en hartweefsel.
De heparan sulfaat proteoglycanen van het
celoppervlak vervullen een essentiële co-factor functie in tal van
receptor-ligand interacties ( groeifactoren, cytokinen ,
adhesiemoleculen, lipoproteïnen , proteïnaseremmers ) die de groei ,
differentiatie , vorm en motiliteit ( het vermogen tot actieve beweging )
van de cellen bepalen.
Chondroïtine sulfaat is een belangrijk
bestanddeel van kraakbeen, zorgt daar voor structuur, houdt het water en
de voedingsstoffen vast en zorgt ervoor dat andere moleculen het kraakbeen
kunnen binnendringen ( een belangrijke functie aangezien het kraakbeen
niet doorbloed is ) . Men treft het aan in bindweefsel, de beenderen , de
pezen en de huid.
Hyaluronzuur is een natuurlijk bestanddeel van
het organisme. Het betreft een polysaccharide macromolecuul, afgescheiden
door sommige cellen van de synovialis ( slijmvliesbekleding van
gewrichtsholten, peesscheden en bursae ) , en samengesteld uit
monosaccharide- eenheden van afwisselend D-glucuronzuur en
N-acetyl-D-glucosamine ( 2-aceetamido-2-desoxy-D-Glucose), die
respectievelijk via β-1,3- en β-1,4 bindingen
aan elkaar gekoppeld zijn. Het moleculair gewicht kan verschillen.
De stof is ruim verspreid in de extracellulaire matrix van het
bindweefsel, in het synoviaal vocht en eveneens in het kamerwater en het
glasvocht van het oog.
Glucosamine is een amino-monosaccharide dat een
rol speelt bij de opbouw en het behoud van kraakbeen. Het dient als
intermediair substraat bij de synthese van de glycosaminoglycanen en de
proteoglycanen van het gewrichtskraakbeen. De stof is op natuurlijke wijze
aanwezig in een groot aantal weefsels en heeft een bijzondere affiniteit
voor kraakbeen.
Verschillende genetisch aangeboren ziekten ,
zoals bijvoorbeeld de lysosomale stapelingsziekten, resulteren uit
defecten van de lysosomale enzymen die verantwoordelijk zijn voor de
stofwisseling van deze complexe membraan-geassocieerde glycosaminoglycanen.
Deze specifieke ziekten , die de mucopolysaccharidosen worden
genoemd ( afkorting MPS ) , leiden tot stapeling van
glycosaminoglycanen in de cel.
Glycoproteïnen:
Glycoproteïne , algemene naam voor een eiwit dat
op één of meer plaatsen een covalent gebonden oligosaccharideketen bevat,
bestaande uit een aantal suikermoleculen.
De suikerketens zijn gebonden aan een zijketen
van een van de aminozuren serine, threonine of asparagine; in bepaalde
eiwitten, zoals bijvoorbeeld collageen , ook aan een hydroxylysine of
hydroxyproline.
In totaal komen in glycoproteïnen negen
verschillende suikers voor, waaronder het neuraminezuur een belangrijke
plaats inneemt. Glycoproteïnen vervullen een groot aantal functies. Zo
dienen zij o.a. als smeermiddel (synovia) voor gewrichten . Veel hormonen
en enzymen zijn eveneens glycoproteïnen. Glycoproteïnen in of op het
plasmamembraan van cellen spelen een belangrijke rol bij de communicatie
tussen cellen onderling en bij de specifieke herkenning van cellen door
moleculen. Voorbeelden zijn : de organisatie van cellen tot weefsels;
herkenning van lichaamsvreemde cellen, zoals bacteriën door antilichamen;
de specifieke herkenning van cellen door hormonen en de bepaling van de
bloedgroep door glycoproteïnen in het plasmamembraan van de rode
bloedcellen.
Stoornissen betreffende de structuur en
degradatie van glycoproteïnen zijn :
alfa-mannosidose, beta-mannosidose, alfa-L-fucosidase deficiëntie,
Sialidose type I en Type II ( neuraminidase deficiëntie ),
aspartylglucosaminurie, Glutamyl Ribose-5-Phosphate Glycoproteinosis en
siaalzuurstapelingsziekte.
Neuraminezuur is een verbinding die ontstaat door
koppeling van pyrodruivenzuur met mannosamine en vormt een belangrijke
bouwsteen van de suikerketens van glycoproteïnen in bacteriële celwanden
en in de "coat" die de buitenkant van het cytoplasmamembraan van dierlijke
cellen bedekt. Verder is het een karakteristiek bestanddeel van bepaalde
in membranen voorkomende sfingolipiden. De NH2-groep van
neuraminezuur is meestal geacetyleerd; men spreekt dan van
N-acetyl-neuraminezuur of sialinezuur. Dit sialinezuur is vaak de
eindstandige suiker van koolhydraatketens van de glycoproteïnen van
celwand en coat. Het speelt daarom een zeer belangrijke rol in de
communicatie van de cel met zijn omgeving die voornamelijk via de
glycoproteïnen tot stand komt. Sialinezuur geeft ook de speciale
eigenschappen aan de mucinen, glycoproteïnen die dienst doen als
smeermiddelen.
Sfingolipiden ( Grieks > sphingoo =
omsluiten en van lipiden )
Sfingolipiden blijken bioactieve moleculen te
zijn en sommige, waaronder sfingosine, ceramide, sfingosine-fosfaat en
ceramide-fosfaat , worden als secundaire boodschappers vooropgesteld.
Sfingolipiden zijn actief betrokken bij de vernieuwing van cellen doordat
ze celgroei en celdood reguleren. Een bepaald soort sfingolipide,
glucosylceramide, speelt een rol in de celdifferentiatie. Dit is het
specialisatieproces waarin een lichaamscel zich aan past aan zijn
specifieke taak. De aan glucosylceramide verwante stof ceramide blijkt
betrokken te zijn bij de regulatie van de ( geprogrammeerde ) celdood.
Ceramiden ( N-acylsfingosinen ) zijn secundaire boodschappers die een rol
spelen in signaal transductie processen. Sfingolipiden spelen belangrijke
rollen in de signaal transductie processen. Stoornissen in de synthese of
afbraak van sfingolipiden zijn o.a. ziekte van Gaucher type I,
Niemann-Pick type A, ziekte van Krabbe en andere.
Sfingolipiden is een heterogene groep lipiden die
gekarakteriseerd zijn door het bezit van het structuuronderdeel sfingosine,
een aminodiol, of van een daarvan afgeleide verbinding. De sfingolipiden
kunnen worden onderverdeeld in drie verschillende groepen :
- Sfingomyelinen, die bij volledige hydrolyse
equimolaire hoeveelheden sfingosine, vetzuur, fosfaat en choline leveren.
Het vetzuur blijkt via een amideverbinding gekoppeld te zijn aan een
2-aminogroep, en het fosforylcholine via een esterbinding aan de
1-hydroxygroep. Vanwege het laatstgenoemde structuurkenmerk behoren
sfingomyelinen tot de fosfolipiden. Sfingomyelinen komen met name in de
zenuwmergscheden voor.
- Cerebrosiden, waarin een vetzuur weer via een
amidebinding gekoppeld is aan de 2-aminogroep, en een suiker, m.n.
galactose, via een glycosidische binding aan de 1-hydroxygroep. In sommige
pathologische gevallen is de galactose grotendeels vervangen door glucose
( bijv. bij de ziekte van Gaucher ). In een aantal gevallen bevindt zich
aan de C-6 van de galactose nog een zwavelzure groep; men spreekt dan van
sulfatiden.
- Gangliosiden : dit zijn zeer complexe
sfingolipiden die opgebouwd zijn uit sfingosine, een vetzuur, 1 of meer
suikers. (vnl. glucose, galactose of N-acetylgalactosamine ). De
cerebrosiden en de gangliosiden worden tezamen ook wel als glycolipiden
aangeduid, gezien het feit dat ze 1 of meer suikers als structuuronderdeel
bezitten.
Glycosfingolipiden zoals gangliosiden worden in
behoorlijke hoeveelheden aangetroffen in zenuwcel membranen.
Fosfolipiden:
Fosfolipiden of fosfatiden zijn fosfaat
bevattende vetachtige stoffen die in alle levende cellen worden
aangetroffen als bestanddeel van de verschillende celmembranen; vooral
zenuwweefsel is rijk aan fosfolipiden. fosfolipiden kunnen worden
onderverdeeld in
- fosfoglyceriden : deze stoffen met als
basiscomponent een fosfatidinezuur, bestaande uit glycerol, veresterd met
twee vetzuurgroepen en een fosforzuurgroep. Deze fosfatidinezuren komen
als zodanig voor in planten , maar in dierlijk weefsel overheersen
fosfatidinezuren waarin een teede OH-groep van het fosforzuur is veresterd
: a. bij lecithinen, b. bij kefalinen of met serine. Cardiolipine (
difosfatidylglycerol ) bestaat uit twee fosfatidinezuurgroepen gekoppeld
via propaantriol; c. bij inositiden met inositol.
- sfingomyelinen : deze stoffen bestaan uit de
aminodialcohol sfingosine, met daaraan gekoppeld een vetzuur ( aan de NH2-groep)
en een fosforylcholine aan de eindstandige OH-groep. Sfingomyelinen komen
met name in de zenuwmergscheden voor.
Lysosomale enzymdeficiënties ( lysosomale stapelingsziekten ):
Een lysosomale enzymdeficiëntie is een
onvoldoende of afwezige werking van in de lysosomen gelokaliseerde
enzymen, waardoor stapeling van stofwisselingsproducten optreedt. Anders
gezegd: wanneer één enzym defect is, dan kan een binnenkomende stof niet
in het lysosoom omgezet worden in een andere stof en zal in het lysosoom
achterblijven waardoor er langzaam maar zeker een stapeling van deze stof
in het lysosoom ontstaat.
Dit bemoeilijkt op den duur het functioneren van
het lysosoom en uiteindelijk het functioneren van de cel.
Hierdoor ontstaan ziektebeelden die bekend staan
als de lysosomale stapelingsziekten.
Beperkt overzicht lysosomale enzymen met
bijbehorend genetisch defect :
Stoornissen betreffende de lysosomale enzymen:
De Lysosomale stapelingsziekten
Glycogen Storage
Disease Type II:
Glycogeen
Stapelingsziekte Type II:
Acid
a-Glucosidase Deficiency ( Acid Maltase )
Deficiency:
-
Glycogen Storage Disease II ( Ziekte van Pompe )
Synoniemen: Acid Alpha Glucosidase Deficiency; GAA Deficiency; Pompe
Disease; Cardiac Form of Generalized Glycogenosis; Cardiomegalia
Glycogenica Diffusa; Acid Maltase Deficiency
( AMD ); Alpha-1,4-glucosidase Deficiency
OMIM:
232300
OMIM:
Clinical Synopsis
e-Medicine:
Glycogen Storage Disease Type II
Extra informatie :
Pompe's Disease Page ;
Acid Maltase
Deficiency Association ;
Pompe Center: Erasmus MC
ExPASy: Alpha-glucosidase
EC 3.2.1.20
Enzym synoniemen: Maltase; Glucoinvertase; Glucosidosucrase;
Maltase-glucoamylase; Lysosomal alpha-glucosidase. Acid maltase.
Hydrolysis of terminal, non-reducing 1,4-linked D-glucose residues with
release of D-glucose.
De Mucopolysaccharidosen:
( The Mucopolysaccharidoses )
-
Mucopolysaccharidosis Type I ( Ziekte van Hurler )
Synoniemen: MPSI; MPS I; Alpha-L-Iduronidase Deficiency; IDA
Deficiency; IDUA Deficiency; Hurler Syndrome.
OMIM:
252800
OMIM:
Clinical Synopsis
Who Named It?:
Gertrud Hurler
e-Medicine:
Mucopolysaccharidosis Type I H
ExPASy: L-iduronidase
EC
3.2.1.76
Hydrolysis of alpha-L-iduronosidic linkages in desulfated dermatan
-
Mucopolysaccharidosis Type I S ( Ziekte van Scheie )
Synoniemen: MPS I S; Scheie Syndrome
OMIM:
252800
OMIM:
Clinical Synopsis
Who Named It?:
Harold Glendon Scheie
e-Medicine:
Mucopolysaccharidosis Type I S
ExPASy: L-iduronidase
EC
3.2.1.76
Hydrolysis of alpha-L-iduronosidic linkages in desulfated dermatan
-
Mucopolysaccharidosis Type I H/S ( Ziekte van Hurler-Scheie )
Synoniemen: MPS I H/S
OMIM:
252800
OMIM:
Clinical Synopsis
Who Named It?:
Gertrud Hurler
Who Named It?:
Harold Glendon Scheie
e-Medicine:
Mucopolysaccharidosis Type I H/S
ExPASy: L-iduronidase
EC 3.2.1.76
Hydrolysis of alpha-L-iduronosidic linkages in desulfated dermatan
-
Mucopolysaccharidosis Type II ( MPS IIA-severe ) ( Ziekte van Hunter )
Synoniemen: Hunter Syndrome; Iduronate-2-sulfatase
deficiency; MPS II; MPS2; IDS Deficiency; Sulfoiduronate
sulfatase Deficiency; SIDS Deficiency
OMIM:
309900
OMIM:
Clinical Synopsis
Who Named It?:
Charles A. Hunter
e-Medicine:
Mucopolysaccharidosis Type II
ExPASy:
Iduronate-2-sulfatase.
EC 3.1.6.13
Enzym synoniemen: Chondroitinsulfatase
Hydrolysis of the 2-sulfate groups of the
L-iduronate 2-sulfate units of dermatan sulfate, heparan sulfate and
heparin
-
Mucopolysaccharidosis Type II ( MPS IIB- mild )
OMIM:
309900
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis Type II
ExPASy:
Iduronate-2-sulfatase.
EC 3.1.6.13
Enzym synoniemen: Chondroitinsulfatase
Hydrolysis of the 2-sulfate groups of the L-iduronate 2-sulfate units of
dermatan sulfate, heparan sulfate and heparin
-
Mucopolysaccharidosis Type IIIA ( Ziekte van Sanfillipo type
A )
Synoniemen: MPSIIIA; MPS3A;
Sanfilippo Syndrome
A; Heparan Sulfate Sulfatase Deficiency; Sulfamidase Deficiency
OMIM:
252900
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis Type III
ExPASy: N-sulfoglucosamine sulfohydrolase
EC 3.10.1.1
Enzym synoniemen: Sulfoglucosamine sulfamidase; Sulphamidase.
N-sulfo-D-glucosamine + H2O <=> D-glucosamine + sulfate
-
Mucopolysaccharidosis Type IIIB (
Ziekte van Sanfilippo type B )
Synoniemen:
Sanfilippo Syndrome B;
N-Acetyl-Alpha-D-Glucosaminidase Deficiency; MPSIIIB;
MPS3B; NAGLU Deficiency
OMIM:
252920
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis Type III
ExPASy: Alpha-N-acetylglucosaminidase
EC 3.2.1.50
Enzym synoniemen: N-acetyl-alpha-glucosaminidase; NAG.
Hydrolyzes UDP-N-acetylglucosamine
-
Mucopolysaccharidosis Type IIIC ( Ziekte van Sanfilippo type
C )
Synoniemen:
Sanfilippo Syndrome C;
Acetyl CoA: Alpha-Glucosaminide
N-Acetyltransferase Deficiency; MPSIIIC; MPS3C
OMIM:
252930
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis Type III
ExPASy: Heparan-alpha-glucosaminide N-acetyltransferase
EC 2.3.1.78
Acetyl-CoA + heparan alpha-D-glucosaminide <=> CoA + heparan
N-acetyl-alpha-D-glucosaminide
-
Mucopolysaccharidosis Type IIID ( Ziekte van
Sanfilippo type D )
Synoniemen: Sanfilippo
Syndrome D;
N-Acetylglucosamine-6-sulfatate sulfatase
deficiency; MPSIIID
OMIM:
252940
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis Type III
ExPASy: N-acetylglucosamine-6-sulfatase
EC 3.1.6.14
Enzym synoniemen: Glucosamine-6-sulfatase; Chondroitinsulfatase
Hydrolysis of the 6-sulfate group of the N-acetyl-D-glucosamine
6-sulfate units of heparan sulfate and keratan sulfate.
-
Mucopolysaccharidosis Type IVA ( Ziekte
van Morquio type A )
Synoniemen: Morquio
Syndrome
A ;
Galactosamine-6-sulfatase deficiency;
MPSIVA; MPS4A; GALNS Deficiency
OMIM:
253000
OMIM:
Clinical Synopsis
Who Named It?:
Luís Morquio
e-Medicine:
Mucopolysaccharidosis Type IV
Extra informatie:
Vereniging Morquio
ExPASy:
N-acetylgalactosamine-6-sulfatase
EC 3.1.6.4
Enzym synoniemen: Chondroitinsulfatase; Chondroitinase;
Galactose-6-sulfate sulfatase
Hydrolysis of the 6-sulfate groups of the N-acetyl-D-galactosamine
6-sulfate units of chondroitin sulfate and of theD-galactose 6-sulfate
units of keratan sulfate
-
Mucopolysaccharidosis Type IVB ( Ziekte van
Morquio type B )
Synoniemen:
Morquio
Syndrome
B
;
Beta-Galactosidase
Deficiency; MPSIVB; MPS4B
OMIM:
253010
OMIM:
Clinical Synopsis
Who Named It?:
Luís Morquio
e-Medicine:
Mucopolysaccharidosis Type IV
Extra informatie:
Vereniging Morquio
ExPASy:
Beta-galactosidase
EC 3.2.1.23
Enzym synoniemen: Lactase; Exo-(1->4)-beta-D-galactanase
Hydrolysis of terminal, non-reducing beta-D-galactose residues in
beta-D-galactosides
-
Mucopolysaccharidosis Type V wordt
niet langer gebruikt.
-
Mucopolysaccharidosis Type VI (
Ziekte van Maroteaux-Lamy )
Synoniemen: Maroteaux-Lamy Syndrome; Arylsulfatase B
Deficiency ; MPSVI; MPS6; ARSB Deficiency;N-Acetylgalactosamine-4-sulfatase
deficiency
OMIM:
253200
OMIM:
Clinical Synopsis
Who Named It?:
Pierre Maroteaux ;
Maurice Emile Joseph Lamy
e-Medicine:
Mucopolysaccharidosis Type VI
ExPASy:
N-acetylgalactosamine-4-sulfatase
EC 3.1.6.12
Enzym synoniemen: Arylsulfatase B; Chondroitinsulfatase; Chondroitinase.
Hydrolysis of the 4-sulfate groups of the N-acetyl-D-galactosamine
4-sulfate units of chondroitin sulfate and dermatan sulfate.
-
Mucopolysaccharidosis Type VII ( Ziekte van Sly )
Synoniemen: Sly Syndrome;
beta-glucuronidase deficiency ; MPSVII; MPS7; GUSB Deficiency
OMIM:
253220
OMIM:
Clinical Synopsis
e-Medicine:
Mucopolysaccharidosis
Type VII
ExPASy:
Beta-glucuronidase
EC 3.2.1.31
A beta-D-glucuronoside + H2O <=> an alcohol +
D-glucuronate
-
Mucopolysaccharidosis Type VIII
wordt niet langer gebruikt.
Pycnodysostosis:
Cathepsin K
deficiency
|
|
- Pycnodysostosis
OMIM:
265800
OMIM:
Clinical Synopsis
Ziekten betreffende de
fosforylisatie en lokalisatie van Lysosomale enzymen:
I-Cell Disease and Pseudo-Hurler
Polydystrofy:
(
Mucolipidose type II en type III )
-
Mucolipidosis II ( Mucolipidose type II )
( I-Cell disease )
Synoniemen:
I-Cell disease; ICD; MLII;
ML2; N-Acetylglucosamine-1-phosphotransferase deficiency; GNPTA
deficiency
OMIM:
252500
OMIM:
Clinical Synopsis
e-Medicine:
I-Cell Disease (Mucolipidosis Type II)
Extra informatie:
I-Cell disease
ExPASy: UDP-N-acetylglucosamine--lysosomal-enzyme
N-acetylglucosaminephosphotransferase
EC 2.7.8.17
UDP-N-acetyl-D-glucosamine + lysosomal-enzyme D-mannose <=> UMP +
lysosomal-enzyme N-acetyl-D-glucosaminyl-phospho-D-mannose
-
Mucolipidosis III (
Mucolipidose type III )
Synoniemen:
Pseudo-Hurler
Polydystrofie; ML III; ML3
OMIM:
252600
OMIM:
Clinical Synopsis
a-N-Acetylgalactosaminidase
Deficiency:
Schindler Disease
( Ziekte van Schindler )
- Schindler Disease
OMIM:
104170
OMIM:
Clinical Synopsis:
ExPASY: Alpha-N-acetylgalactosaminidase
EC 3.2.1.49
Enzym synoniemen: Alpha-galactosidase B. Alpha-NAGA.
Splits N-acetylgalactosaminyl groups from O-3 of Ser and Thr.
- Schindler Disease Type II
Ziekten betreffende
de structuur en degradatie van Glycoproteïnen:
- Lysosomal Alpha B Mannosidosis (
a-mannosidose)
Synoniemen: Alpha Mannosidosis;
Lysosomal Alpha-D-Mannosidase Deficiency; Alpha Mannosidase B Deficiency
OMIM:
248500
OMIM:
Clinical Synopsis
ExPASy: Alpha-mannosidase
EC 3.2.1.24
Hydrolysis of terminal, non-reducing alpha-D-mannose residues in
alpha-D-mannosides
- Lysosomal Beta A Mannosidosis (
b-mannosidose
)
Synoniemen: Bèta Mannosidosis (
MANB1 ); Beta Mannosidase Deficiency; Beta Mannosidosis
OMIM:
248510
OMIM:
Clinical Synopsis
ExPASy: Beta-mannosidase
EC 3.2.1.25
Enzym synoniemen: Mannanase; Mannase
Hydrolysis of terminal, non-reducing beta-D-mannose residues in
beta-D-mannosides.
-
Fucosidosis ( Fucosidose )
Synoniemen: Alfa -L-fucosidase
deficiency
OMIM:
230000
OMIM:
Clinical Synopsis
ExPASy: Alpha-L-fucosidase
EC 3.2.1.51
Enzym synoniemen: Alpha-L-fucoside fucohydrolase
een alpha-L-fucoside + H2O <=> een alcohol + L-fucose
-
Neuraminidase deficiency
( neuraminidase deficiëntie
)
Synoniemen: Sialidosis Types I and II; Cherry Red Spot and
Myoclonus Syndrome; Sialidase Deficiency;
NEUI deficiency; Glycoprotein
Neuraminidase deficiency; NEUG Deficiency; Neuraminidase I Deficiency;
NEU Deficiency.
OMIM:
256550
OMIM:
Clinical Synopsis
ExPASy: Exo-alpha-sialidase
EC 3.2.1.18
Enzym synoniemen; Sialidase. Neuraminidase. N-acylneuraminate
glycohydrolase. Alpha-neuraminidase
Hydrolysis of alpha-(2->3)-, alpha-(2->6)-, alpha-(2->8)-glycosidic
linkages of terminal sialic residues in oligosaccharides, glycoproteins,
glycolipids, colominic acid and synthetic substrates
-
Aspartylglucosaminuria
(
aspartylglucosaminurie )
Synoniemen: Glycosylasparaginase deficiency ; Aspartylglucosaminidase
Deficiency; AGU; AGA Deficiency; Glycoasparaginase;Aspartylglycosaminuria
OMIM:
208400
OMIM:
Clinical Synopsis
ExPASy: N(4)-(beta-N-acetylglucosaminyl)-L-asparaginase
EC 3.5.1.26
Enzym synoniemen: Aspartylglucosylamine deaspartylase.
Aspartylglucosylaminidase. Glycosylasparaginase
N4-(beta-N-acetyl-D-glucosaminyl)-L-asparagine + H2O <=>
N-acetyl-beta-glucosaminylamine + L-aspartate
-
Glutamyl Ribose-5-Phosphate Storage Disease
Synoniemen: ADP-Ribose Protein Hydrolase Deficiency
OMIM:
306920
-
Infantile Sialic Acid Storage Disorder
(
Siaalzuur
stapelingsziekte )
Synoniemen: ISSD; Sialuria ( infantile
form ); N-Acetylneuraminic acid storage disease; NANA storage disease;
NSD.
OMIM:
269920
OMIM:
Clinical Synopsis
OMIM:
Sialin
- Sialuria, Finnish type
Synoniemen: Salla disease
OMIM:
604369
OMIM:
Clinical Synopsis
Acid Lipase
Deficiency:
- Wolman
Disease ( severe infantile onset )
Synoniemen: Lysosomal Acid Lipase Deficiency;
LIPA Deficiency; LAL Deficiency; Acid Cholesteryl Ester Hydrolase
Deficiency; Cholesterol ester hydrolase deficiency; Cholesterol ester
storage disease; Cholesteryl ester storage disease; Acid cholesteryl
ester hydrolase deficiency, type II.
OMIM:
278000
OMIM:
Clinical Synopsis
ExPASy: Sterol esterase
EC 3.1.1.13
Enzym synoniemen: Cholesterol esterase. Cholesterol ester synthase.
Triterpenol esterase
A steryl ester + H2O <=> a sterol a fatty acid
-
Cholesteryl Ester Storage Disease (
mild late onset ) (
Cholesteryl ester stapelingsziekte )
OMIM:
278000
OMIM:
Clinical Synopsis
ExPASy: Sterol esterase
EC 3.1.1.13
Enzym synoniemen: Cholesterol esterase.Cholesterol ester synthase.
Triterpenol esterase
A steryl ester + H2O <=> a sterol a fatty acid
Ceramidase Deficiency: Farber Lipogranulomatosis:
( Ziekte van Farber )
-
Farber Lipogranulomatosis
Synoniemen: Farber Disease; Ceramidase deficiency; Acid ceramidase
deficiency; AC deficiency; N-Laurylsphingosine deacylase deficiency.
OMIM:
228000
OMIM:
Clinical Synopsis
Who Named It?:
Sidney Farber
ExPASy: Ceramidase
EC 3.5.1.23
Enzym synoniemen: Acylsphingosine deacylase
N-acylsphingosine + H2O <=> a fatty acid + sphingosine
Niemann-Pick
Diseases Types A and B:
Acid Sphingomyelinase Deficiencies:
-
Niemann-Pick Disease Type A, B and E
Synoniemen: Sphingomyelin lipidosis; Sphingomyelinase deficiency
OMIM:
257200
OMIM:
Clinical Synopsis
Who Named It?:
Albert Niemann ;
Ludwig Pick
e-Medicine:
Niemann-Pick Disease
ExPASy: Sphingomyelin phosphodiesterase
EC 3.1.4.12
Enzymsynoniemen: Neutral sphingomyelinase; Acid sphingomyelinase;
Sphingomyelin + H2O <=>
N-acylsphingosine + choline phosphate
Niemann-Pick
Disease Type C:
A Cellular Cholesterol Lipidosis:
( Ziekte van Niemann-Pick type C )
-
Niemann-Pick Disease Type C
Synoniemen: NPC; Niemann-Pick Diease Type C1; Niemann-Pick disease
with cholesterol esterification block; Niemann-Pick disease, subacute
juvenile form; Niemann-Pick disease, chronic neuropathic form.
OMIM:
257220
OMIM:
Clinical Synopsis
Who Named It?:
Albert Niemann ;
Ludwig Pick
-
Niemann-Pick Disease Type C2
OMIM:
601015
Who Named It?:
Albert Niemann ;
Ludwig Pick
Niemann-Pick disease without
sphingomyelinase deficiency:
- Niemann-Pick disease
without sphingomyelinase deficiency
Synoniemen: Niemann-Pick disease, Nova Scotian type; Niemann-Pick
Disease Type D
OMIM:
257250 moved to
257220
Who Named It?:
Albert Niemann ;
Ludwig Pick
Gaucher Disease:
( Ziekte van
Gaucher )
Extra informatie: Gaucher
Vereniging Nederland
Extra informatie: The
National Gaucher Foundation
Extra informatie: Gauchers
Association
Extra informatie:
Gaucher Disease ( pdf-document: hier heeft u Acrobat Reader voor
nodig )
- Gaucher Disease, Type I
Synoniemen: GD I;
Glucocerebrosidase Deficiency; Gaucher disease, Noncerebral
Juvenile;
Acid
Beta-Glucosidase Deficiency;GBA
Deficiency.
OMIM:
230800
OMIM:
Clinical Synopsis
e-Medicine:
Gaucher Disease ;
Gaucher Vereniging
Nederland
ExPASy:
Glucosylceramidase
EC 3.2.1.45
Enzym synoniemen:
Beta-glucocerebrosidase. Acid beta-glucosidase.
D-glucosyl-N-acylsphingosine glucohydrolase.
D-glucosyl-N-acylsphingosine + H2O <=> D-glucose +
N-acylsphingosine
- Gaucher Disease, Type II
Synoniemen: GD II;
Gaucher Disease, Infantile Cerebral; Gaucher Disease, Acute
Neuronopathic type.
OMIM:
230900
OMIM:
Clinical Synopsis
e-Medicine:
Gaucher Disease ;
Gaucher Vereniging
Nederland
ExPASy:
Glucosylceramidase
EC 3.2.1.45
Enzym synoniemen:
Beta-glucocerebrosidase. Acid beta-glucosidase.
D-glucosyl-N-acylsphingosine glucohydrolase.
D-glucosyl-N-acylsphingosine + H2O <=> D-glucose +
N-acylsphingosine
- Gaucher Disease, Type
III
Synoniemen: GD III; Gaucher Disease, juvenile and adult, cerebral;
Gaucher Disease, chronic neuronopathic type; Gaucher Disease, subacute
neuronopathic type.
OMIM:
231000
OMIM:
Clinical Synopsis
e-Medicine:
Gaucher Disease ;
Gaucher Vereniging
Nederland
ExPASy:
Glucosylceramidase
EC 3.2.1.45
Enzym synoniemen:
Beta-glucocerebrosidase. Acid beta-glucosidase.
D-glucosyl-N-acylsphingosine glucohydrolase.
D-glucosyl-N-acylsphingosine + H2O <=> D-glucose +
N-acylsphingosine
Galactosylceramide
Lipidosis:
Globoid-Cell
Leukodystrophy
Krabbe Disease
( Ziekte van
Krabbe )
Extra informatie :
Ziekte van Krabbe: vroege infantiele vorm
Extra informatie:
Haley's Hope
- Krabbe Disease
Synoniemen: Globoid-cell leukodystrophy; GLD; GCL; Globoid-cell
leukoencephalopathy; Galactocerebrosidase deficiency;Galactocylceramide
beta-galactosidase deficiency.
OMIM:
245200
OMIM:
Clinical Synopsis
Who Named It?:
Knud Haraldsen Krabbe
e-Medicine:
Krabbe Disease
ExPASy: Galactosylceramidase
EC 3.2.1.46
Enzym synoniemen: Galactocerebrosidase. Galcerase. Galactosylceramide
beta-galactosidase. Galactocerebroside beta-galactosidase
D-galactosyl-N-acylsphingosine + H2O <=>D-galactose +
N-acylsphingosine
Metachromatic leukodystrophy
and
Multiple Sulfatase Deficiency:
Sulfatide Lipidosis
Extra informatie:
Metachromatische leukodystrofie
- Metachromatic Leukodystrophy
( metachromatische leukodystrofie )
Synoniemen: MLD; Metachromatische
Leukoencephalopathie; Sulfatide Lipidose;
Arylsulfatase A Deficiëntie
;ARSA Deficiëntie;
Cerebroside Sulfatase Deficiëntie
OMIM:
250100
OMIM:
Clinical Synopsis
e-Medicine:
Metachromatic Leukodystrophy
ExPASy: Cerebroside-sulfatase
EC 3.1.6.8
Enzym synoniemen: Arylsulfatase A.
A cerebroside 3-sulfate + H2O <=> a cerebroside + sulfate
- Multiple Sulfatase Deficiency
Synoniemen: MSD; Mucosulfatidosis; Juvenile sulfatidosis, Austin Type.
OMIM:
272200
OMIM:
Clinical Synopsis
ExPASy: Human Arylsulfatase and Glycosaminoglycan Sulfatases involved
with Multiple Sulfatase Deficiency:
Arylsulfatase A
EC 3.1.6.8
Arylsulfatase B
EC 3.1.6.12
Arylsulfatase C
EC 3.1.6.2
L-iduronidase
EC
3.2.1.76
N-sulfoglucosamine sulfohydrolase
EC 3.10.1.1
N-acetylgalactosamine-6-sulfatase
EC 3.1.6.4
N-acetylglucosamine-6-sulfatase
EC 3.1.6.14
a-Galactosidase A Deficiency:
Fabry Disease
( Ziekte van
Fabry )
Extra informatie:
Fabry Support en Informatie
Groep Nederland
-
Fabry Disease
Synoniemen: Diffuse Angiokeratoma; Anderson-Fabry Disease; Hereditary
Distopic Lipidosis; Alpha Galactosidase A deficiency;GLA Deficiency;
Ceramide Trihexosidase Deficiency.
OMIM:
301500
OMIM:
Clinical Synopsis
Who Named It?:
Johannes Fabry
e-Medicine:
Fabry Disease
e-Medicine:
New
Therapies
ExPASy: Alpha-galactosidase
EC 3.2.1.22
Enzym synoniemen: Melibiase
Melibiose + H2O <=> galactose + glucose
b-Galactosidase
Deficiency ( b-Galactosidosis ):
Gm1
Gangliosidosis and Morquio B Disease
- Gangliosidosis, generalized
Gm1, Type I
Synoniemen: Beta Galactosidase 1 Deficiency; GLB1 Deficiency
OMIM:
230500
OMIM:
Clinical Synopsis
e-Medicine:
GM1
Gangliosidosis
ExPASy: Beta-galactosidase
EC 3.2.1.23
Enzym synoniemen: Lactase; Exo-(1->4)-beta-D-galactanase
Hydrolysis of terminal, non-reducing beta-D-galactose residues in
beta-D-galactosides.
- Gangliosidosis, generalized
Gm1, Type II, or juvenile type
OMIM:
230600
OMIM:
Clinical Synopsis
- Gangliosidosis, generalized
Gm1, Type III, or adult type
OMIM:
230650
OMIM:
Clinical Synopsis
- Morquio Disease, Type B
OMIM:
253010
OMIM:
Clinical Synopsis
Who Named It?:
Luís Morquio
e-Medicine:
Mucopolysaccharidosis Type IV
Extra informatie:
Vereniging Morquio
ExPASy:
Beta-galactosidase
EC 3.2.1.23
Enzym synoniemen: Lactase; Exo-(1->4)-beta-D-galactanase
Hydrolysis of terminal, non-reducing beta-D-galactose residues in
beta-D-galactosides
Galactosialidosis
- Neuraminidase deficiency
with beta-galactosidase deficiency
Synoniemen: Goldberg syndrome; Galactosialidosis; GSL; Neuraminidase
/ beta-galactosidase expresion; NGBE; Deficiency of lysosomal
protective protein; Deficiency of cathepsin A; Protective protein/cathepsin
A deficiency; PPCA deficiency.
OMIM:
256540
OMIM:
Clinical Synopsis
ExPASy:
EC 3.4.16.5 Carboxypeptidase C
Synoniemen voor dit enzym: Serine-type carboxypeptidase I. Cathepsin A.
Carboxypeptidase Y.
Lysosomal protective protein
The Gm2
Gangliosidoses
- Tay-Sachs Disease ( TSD )
( Ziekte van Tay-Sachs )
Synoniemen: Gm2 Gangliosidosis type I; B variant GM2
Gangliosidosis; Hexosaminidase A deficiency; HEXA Deficiency.
OMIM:
272800
OMIM:
Clinical Synopsis
OMIM:
Hexosaminidase A; HEXA
Who Named It?:
Warren Tay ;
Bernard Sachs
ExPASy: Beta-N-acetylhexosaminidase
EC 3.2.1.52
Enzym synoniemen: Beta-hexosaminidase; Hexosaminidase;
N-acetyl-beta-glucosaminidase
Hydrolysis of terminal non-reducing N-acetyl-D-hexosamine residues in
N-acetyl-beta-D-hexosaminides.
- Sandhoff Disease (
Ziekte van Sandhoff )
Synoniemen: GM2 Gangliosidosis Type II; Hexosaminidases A and B
Deficiency.
OMIM:
268800
OMIM:
Clinical Synopsis
OMIM:
Hexosaminidase B; HEXB
ExPASy: Beta-N-acetylhexosaminidase
EC 3.2.1.52
Enzym synoniemen: Beta-hexosaminidase; Hexosaminidase;
N-acetyl-beta-glucosaminidase
Hydrolysis of terminal non-reducing
N-acetyl-D-hexosamine residues in N-acetyl-beta-D-hexosaminides.
- Tay-Sachs Disease, AB variant
Synoniemen: Hexosaminidase activator deficiency; GM2 gangliodidosis
type AB; AB variant GM2 gangliosidosis.
OMIM:
272750
OMIM:
Clinical Synopsis
Who Named It?:
Warren Tay ;
Bernard Sachs
- Gangliosidosis, GM2, Juvenile,
A( M )B variant
OMIM:
230710
OMIM:
Clinical Synopsis
Lysosomale
transport stoornissen
- Cystinosis, nephropathic; CTNS
( infantiele cystinose )
Synoniemen: Defect of lysosomal cystine transport protein; Defect of
cystinosin.
OMIM:
219800
OMIM:
Clinical Synopsis
OMIM:
Cystinosin
- Cystinosis, late-onset
juvenile or adolescent nephropathic type ( Juveniele cystinose )
OMIM:
219900
OMIM:
Clinical Synopsis
OMIM:
Cystinosin
- Cystinosis, adult
nonnephropathic ( Volwassen cystinose )
Synoniemen: Cystinosis, ocular nonnephropathic; Cystinosis, benign
nonnephropathic.
OMIM:
219750
OMIM:
Clinical Synopsis
OMIM:
Cystinosis
- Sialuria, Finish type
Synoniemen: Salla Disease; SD.
OMIM:
604369
OMIM:
Clinical Synopsis
-
Infantile Sialic Acid Storage Disorder
(
Siaalzuur stapelingsziekte )
Synoniemen: ISSD; Sialuria ( infantile form ); N-Acetylneuraminic
acid storage disease; NANA storage disease; NSD.
OMIM:
269920
OMIM:
Clinical Synopsis
OMIM:
Sialin
Neuronal
Ceroid Lipofuscinoses
e-Medicine: Neuronal
Ceroid Lipofuscinoses
GeneReviews:
Neuronal
Ceroid Lipofuscinoses
1
Ceroid Lipofuscinosis, neuronal, dominant, Parry Type
2
Ceroid Lipofuscinosis, neuronal 1, infantile
( Santavuori of
Santavuori-Haltia Disease )
3
Ceroid Lipofuscinosis, neuronal 2, late infantile ( Jansky
Bielschowsky Disease )
4
Ceroid Lipofuscinosis, neuronal 3, juvenile, ( Vogt-Spielmeyer
Disease )
5
Ceroid Lipofuscinosis, neuronal 4 ( Kufs Disease )
6
Ceroid Lipofuscinosis, neuronal 5
7
Ceroid Lipofuscinosis, juvenile neuronal, with granular osmiophilic
deposits
8
Ceroid Lipofuscinosis, neuronal 6, late infantile, variant
9
Ceroid Lipofuscinosis, neuronal, 8
 |
Elektron Microscopic Atlas :
Lysosomen |
 |
Hoofdmenu |
|
