Mitochondriën
|
|
|
|
|
Mitochondriën
|
|
|
|
|
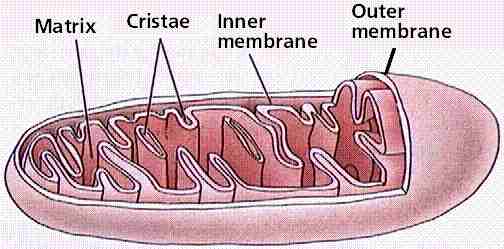
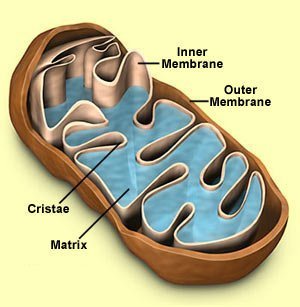
Mitochondriën:Enkelvoud: mitochondrion ( Grieks van "mito" en "khóndrion" ).Ook wel: mitochondrium ( verlatijnste vorm), meervoud is dan mitochondriën of mitochondria. Mitochondriën zijn celorganellen die de in koolhydraten en vetten aanwezige energie overdragen aan ATP en zo ter beschikking stellen van energievragende reacties in de cel. Het mitochondrion werd in 1886 voor het eerst gezien door Richard Altmann (Duits histoloog; 1852- Leipzig 1901), die dacht dat deze organellen intracellulaire parasieten waren. (Hij observeerde verschillende granula in het cytoplasma van diverse cellen. Hij dacht dat deze granula "elementaire organismen" konden zijn die in het cytoplasma leefden en noemde deze "bioblasts"). In 1897 vormde Carl Benda ( geboren op 30-12-1857 in Berlijn en overleden op 24-05-1932 in Turijn ) het woord mitochondrion uit " mito " ( schroefdraad) en " khóndrion " ( korreltje ), omdat deze organellen onder een lichtmicroscoop op schroefdraadvormige korreltjes lijken.
Korte historie:
Historie:Wetenschappers geloven dat ongeveer 1,5 miljard jaar geleden eukaryote cellen hun benodigde energie verkregen door een serie relatief inefficiënte reacties, waarbij geen zuurstof nodig was. Zuurstof, dat een afvalproduct van enkele van deze processen was, begon zich langzaam in de atmosfeer op te bouwen. Wetenschappers denken dat tijdens deze periode een primitieve eukaryote cel een primitieve bacterie heeft verslonden die het vermogen had gekregen zuurstof te gebruiken om grote hoeveelheden energie te produceren. Over miljoenen jaren begon er een symbiotische relatie te ontstaan tussen de cellen en vandaag de dag hebben alle cellen van planten en dieren organellen die de afstammelingen zijn van deze oorspronkelijke energie fabriekjes. In dierlijke cellen worden deze organellen mitochondriën genoemd. Planten hebben naast mitochondriën nog een tweede soort energie producerend organel, namelijk de chloroplast.
De endosymbiose theorie:
De intracellulaire organisatie van de levende cel , bestaande uit
gespecialiseerde organellen, maken complexe levensvormen mogelijk.
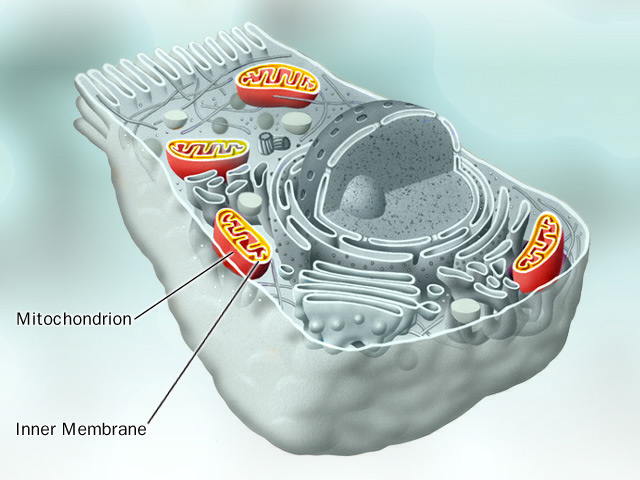
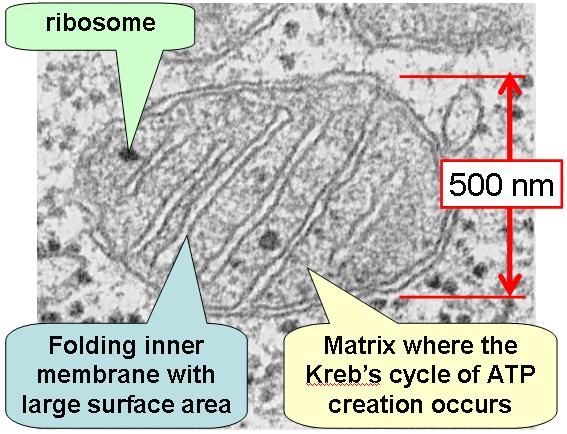
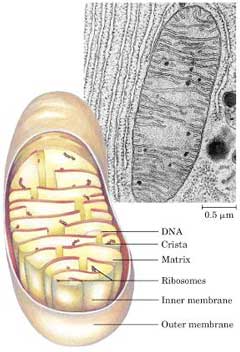
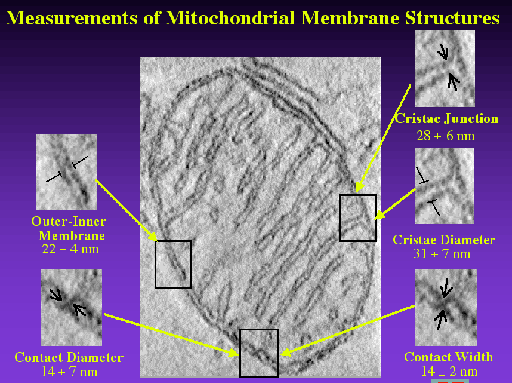
In het hierboven gestelde scenario was de aërobische bacterie die door de anaërobische bacterie was ingenomen , een proto mitochondrion. Met andere woorden , een organisme die het mogelijk maakte energie te produceren uit zuurstof. Symbiose , de relatie waarbij beide soorten een gezamenlijk voordeel behalen, zoals hierboven beschreven, wordt door sommigen beschouwd als het proces waarbij mitochondriën organellen werden in eukaryote cellen. Voordat we verder gaan graven in het bewijs voor deze theorie, moeten we eerst eens kijken naar de functie en structuur van de mitochondriën zoals we die vandaag de dag in onze cellen aantreffen. Mitochondriën zijn eukaryote organellen die de oxidatieve ademhaling uitvoeren, de laatste stap in de cellulaire ademhaling. De oxidatieve ademhaling breekt pyruvaat af die in de glycolyse wordt gevormd waarbij kooldioxide wordt gevormd en produceert de grootste hoeveelheid ATP in de cellen. In eukaryote cellen is zuurstof nodig , omdat mitochondriën zuurstof gebruiken als de uiteindelijke elektronen acceptor in de elektronen transportketen, die tenslotte resulteert in een protonen gradiënt die de ATP synthese aandrijft. Mitochondriën zijn in verschillende hoeveelheden aanwezig in verschillende eukaryote cellen . Cellen die veel energie nodig hebben zoals spierweefsel en de lever hebben verhoudingsgewijs meer mitochondriën dan bijvoorbeeld cellen die minder energie nodig hebben zoals botweefsel. Hoe is het mitochondrion opgebouwd? De kenmerken die we hier bekijken zijn de omvang van het mitochondrion, de membraan structuur, proteïnen status en genetische informatie. Mitochondriën zijn een van de grootste organellen in eukaryote cellen met een omvang van 0,3-1,0 micrometer tot 5-10 micrometer. Het heeft twee membranen waarvan de binnenste sterk geplooid is en cristae worden genoemd. De cristae zijn het startpunt voor enzymen en elektronendragers ( cytochromen ) die verantwoordelijk zijn voor de elektronen transport en de oxidatieve fosforylering. Vergeleken met alle andere organellen in de cel zijn mitochondriën uniek omdat ze hun eigen DNA bevatten, dat wil zeggen , gescheiden van het DNA in de celkern. Enkele proteïnen van het mitochondrion worden door ribosomen in het mitochondrion geproduceerd , overeenkomstig zijn eigen onafhankelijk DNA. Wat is nu het bewijs die de endosymbiose theorie van het mitochondrion en de eukaryote cel ondersteunt? Enkele van de meest overtuigende bewijzen van de symbiose theorie werd hierboven reeds genoemd. Indien mitochondriën eens vrij levende bacteriën waren, bestaat de mogelijkheid dat er overblijfselen van hun vroegere conditie zijn overgebleven , ondanks dat ze vandaag de dag organellen zijn. Hieronder zullen we 6 punten bespreken:
Bijvoorbeeld : cycloheximide blokkeert eukaryote ribosomen door de uitwerking op de overdracht van tRNA, maar het heeft geen uitwerking op mitochondriën en bacteriën. Aan de ander kant zullen stoffen die de prokaryote synthese blokkeren, maar niet de eukaryote synthese, ook de mitochondriële synthese blokkeren, bijvoorbeeld erythromycine en tetracycline (Margulis, 1981, p. 217-218). De structurele overeenkomsten tussen mitochondriën en bacteriën zijn overweldigend , maar zeker niet afdoende. Velen betwijfelen de mogelijkheid van de symbiose theorie …. Dat er een vrij levende proto mitochondrion bestond ten tijde van de opbouw van zuurstof in de atmosfeer …, dat het op de een of andere manier een proto eukaryote cel binnendrong…., dat het een partner was in een symbiotische relatie die uiteindelijk resulteerde in een proto mitochondrion, daarbij zijn autonomie aan een grotere cel gevend en de weg vrij makend voor de eukaryote cel….. wordt dit te veel? Omdat evolutie in vele opzichten de historie is van verschillende chemische reacties van de formatie van de aarde tot de biochemische reacties in levende cellen , zullen we de mogelijkheid van de symbiose theorie eens bekijken. Ten eerste is het niet ondenkbaar dat vrij levende aërobische bacteriën die hoge energie moleculen zoals ATP produceren , een relatie aangaan waarbij die energie kan worden gebruikt. Ten tweede moet de productie van al die energie een enorme input van energie hebben vereist. Bijvoorbeeld een voldoende, beschikbare en efficiënte voedselbron. Ten derde, de komst van zuurstof in de atmosfeer, dat een giftig gas was voor de meerderheid van de organismen op dat moment op aarde, vereiste een nieuwe vorm van stofwisseling , gebaseerd op een nieuwe chemie. Ten slotte zouden proto eukaryote cellen die geen mogelijkheid hadden zuurstof in de atmosfeer om te zetten , moeilijk kunnen overleven. De basis voor de symbiose is duidelijk : anaërobische organismen leverden een constante bron van voedsel en fosfolipiden voor zowel de mitochondriële membranen als voor bacteriën. (Crawford and Marsh, 1995, p. 71-72). Als tegenprestatie stelde de energie die vrijkwam bij de ademhaling van zuurstof, de gast in staat te overleven en zich verder aan te passen aan de nieuwe condities op de aarde. De symbiose theorie wordt gestaafd door natuurlijke , geobserveerde voorbeelden van symbiotische relaties. Bepaalde zeevissen zijn in staat licht te emitteren vanwege de aanwezigheid van lichtgevende bacteriën in hun binnenste. Deze lichtgevende bacteriën leven ook vrijelijk in zeewater, maar geven dan geen licht. (Dyer and Obar, 1985, p. 127). Andere voorbeelden van symbiose zijn de relaties tussen verschillende schimmels en cyanobacteriën, algen en planten en bacteriën en zoogdieren. (Margulis, 1981, p. 165). Hoewel de transformatie van deze symbionten een enorme stap is , als deze al heeft plaatsgevonden, vond deze wel plaats over een periode van miljoenen jaren. Om de hierboven genoemde redenen kan men de theorie, dat mitochondriën evolueerden uit vrij levende aërobische bacteriën tot organellen in de eukaryote cel, niet negeren. De vraag blijft echter , hoe en uit wat konden proto mitochondriën evolueren naast blauw groene algen? Op welke manier drongen de proto mitochondriën de grotere aërobische cel binnen? Konden voorbeelden van vrij levende proto mitochondriën overleven? Bestaan zij of hun nakomelingen vandaag de dag nog? Zijn andere eukaryote organellen ook afgeleid van vrij levende organismen ? Vanwege de bewijzen kan men de symbiose theorie niet negeren. REFERENCE CITATIONS 1. Crawford, Michael and David Marsh, 1995. Nutrition and Evolution, p. 65-83. Keats Publishing, Inc., New Canaan, CT. 2. Dyer, Betsey Dexter and Robert Obar (editors), 1985. The Origin of Eukaryotic Cells, Van Nostrand Reinhold Company, Inc., NY. Papers by: a.) Goksøyr, J., 1967, "Evolution of Eucaryotic Cells;" b.) Schwartz, R.M. and M.O. Dayhoff, 1978, "Origins of Prokaryotes, Eukaryotes, Mitochondria, and Chloroplasts;" c.) Raven, P.H., 1970, "A Multiple Origin for Plastids and Mitochondria;" d.) Doolittle, W.F., 1980, "Revolutionary Concepts in Evolutionary Cell Biology;" and e.) Smith, D.C., 1979, "From Extracellular to Intracellular: The Establishment of a Symbiosis." 3. Margulis, Lynn, 1981. Symbiosis in Cell Evolution, p. 206-227. W. H. Freeman and Company, San Francisco. 4. Prescott, L.M., J.P. Harley and D.A. Klein, 1996. Microbiology, third edition. Wm. C. Brown Publishers, Dubuque, IA
|

|
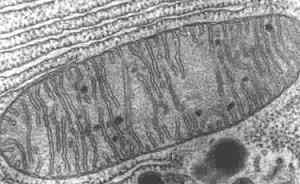
Elektron Microscopic Atlas : Mitochondriën |
Ze hebben een autonoom karakter: ze bevatten DNA, beschikken over
een eigen RNA-synthese en bezitten ribosomen (die sterk
overeenkomen met ribosomen van bacteriën).
Mitochondriën zwellen op en krimpen in als respons op
verschillende hormonen en drugs en tijdens de synthese van ATP.
Dit opzwellen en inkrimpen lijkt gerelateerd te zijn aan de
beweging van water door de cellen.
Mitochondriën kunnen zich, evenals de kern en de plastiden, alleen
vermeerderen door deling. Mitochondriën delen zich op een manier
die morfologisch lijkt op de manier zoals bacteriën zich delen.
De aantallen mitochondriën worden geregeld door autofagie; dit is
een proces waarbij de lysosomen betrokken zijn.
Mitochondriën bevatten o.a. de enzymen van de citroenzuurcyclus en
van de ademhalingsketen (eindoxidatie), inclusief die voor de
vorming van het energierijke ATP en ook die voor de afbraak van
vetzuren.
Een deel van deze enzymen is aan de cristae mitochondriales
gebonden.
Mitochondriën kunnen vele verschillende vormen aannemen en dienen
voor een groot aantal verschillende functies. Elke vorm van het
mitochondrion is karakteristiek voor de cel waarin het
verblijft. Er zijn ongeveer 250 verschillende typen cellen in
ons lichaam.
Er zijn verschillende mitochondriën met specifieke metabole
functies voor veel van de 250 typen cellen in ons
lichaam.
Mitochondriën zijn in staat hun structuur aan te passen om
tegemoet te komen aan veranderende omstandigheden van de cel. Ook
kan er in bepaalde gevallen een toename van de cristae
plaatsvinden, of een verandering van de vorm van het mitochondrion
die resulteert in een groter actief oppervlak voor de omzetting
van energie.
Een mitochondrion kan versmelten met een andere mitochondrion, of
kunnen in grootte toenemen , waarbij " giant mitochondria
" of " megamitochondria " worden
gevormd.
Er wordt een toename van het aantal mitochondriën gegenereerd in
situaties van hoge metabole activiteit.
Aantallen mitochondriën kunnen afnemen door pycnose (
degeneratieve verdichting van de celkern ) , door opzwellen of
door de formatie van autofagolysosomen.
Men denkt dat de levensduur van mitochondriën tussen 5
en 12 dagen ligt.
Voor de biogenese van mitochondriën is het noodzakelijk dat enige
honderden genen gecoördineerd tot expressie komen.
De meeste mitochondriale componenten worden gecodeerd in de kern,
slechts enkele op het mitochondriale DNA.
De meeste cellen met een celkern bevatten in het cytoplasma
( protoplasma of vloeibare gedeelte van de cel)
500 tot 2000 mitochondriën . Sommige cellen hebben maar een paar
mitochondriën. Bloedplaatjes hebben maar twee tot zes
mitochondriën. Rode bloedlichaampjes hebben geen mitochondriën
.Grotere aantallen zijn er voornamelijk te vinden
in de organen in het lichaam die veel energie nodig hebben , zoals
het hart, het brein,de lever ,de skeletspieren, inwendige
klieren en het beenmerg. Dit zijn dan ook de orgaansystemen die in
het algemeen betrokken zijn bij de mitochondriële ziekten.
De gedachte dat mitochondriën alleen als energiefabriekjes voor de
cel fungeren is een misvatting.
Er zijn ongeveer 3000 genen nodig om een mitochondrion te maken.
Het mitochondriële DNA codeert maar voor 37 van deze genen. De
resterende genen worden gecodeerd in de celkern en de resulterende
proteïnen worden naar het mitochondrion getransporteerd.
Enkele algemene kenmerken van het mitochondriële DNA (mtDNA):
Mitochondrial DNA (mtDNA):
General features ( Neuromuscular Disease Center, Washington University,
St. Louis, MO USA )
Maar ongeveer 3 % van de genen die nodig zijn om een mitochondrion
te maken ( 100 van de 3000 ) zijn toegewezen om ATP te
maken. Meer dan 95 % ( 2900 van de 3000 ) zijn betrokken bij
andere functies gebonden aan de speciale taken van de cel waarin
het mitochondrion verblijft. Deze taken veranderen naarmate ons
lichaam zich ontwikkeld van embryo naar volwassene en onze
weefsels groeien en zich aanpassen aan onze omgeving. Deze andere
, niet ATP gerelateerde functies, zijn innig betrokken bij de
meeste belangrijke metabole paden die door de cel gebruikt worden,
om zijn moleculaire bouwsteentjes op te bouwen , af te breken of
te recyclen.
Cellen kunnen zonder mitochondriën geen RNA en DNA maken die ze
nodig hebben om te groeien en te functioneren. De bouwstenen voor
RNA en DNA zijn purinen en pyrimidines.
Mitochondriën bevatten de "rate-limiting" enzymen van de
pyrimidine biosynthese en haem synthese die nodig zijn om
hemoglobine te maken.
Mitochondriën in de lever zijn gespecialiseerd om ammoniak in de
ureum-cyclus
om te zetten. Mitochondriën zijn ook nodig voor de cholesterol
stofwisseling, voor de synthese van testosteron en oestron
en voor de neurotransmitter stofwisseling.
ATP werd in 1929 ontdekt door de Duitse chemicus Karl Lohman.
Synoniemen voor ATP:
Adenosinetrifosfaat
Adenosinetrifosfaat ( ATP ) is een nucleotide ( stof opgebouwd uit
een stikstofrijk molecuul (( purine - of pyrimidine-base )) , een
suikermolecuul en fosforzuur ) dat als co-factor bij vele
enzymatische reacties in het lichaam wordt gebruikt.
ATP wordt uit adenosinedifosfaat ( ADP ) gevormd bij de
oxidatie van suikers, eiwitten en vetten en wordt verbruikt bij
energiebehoevende processen in de cel.
ATP wordt beschouwd als een zgn. energierijke verbinding, omdat
bij de hydrolyse van de trifosfaatgroep energie vrijkomt die voor
andere doeleinden in het metabolisme gebruikt kunnen worden.
Daardoor is deze verbinding geschikt om energie te leveren bij de
spiercontractie, actief transport , signaalversterking, om
proteïnen te synthetiseren, of voor het kopiëren van DNA, en de
opbouw van nieuwe organellen en het pompen van water door
membranen en de verplaatsing van cellen.
De hydrolyse van de fosfaatanhydridefunctie in ATP is een
thermodynamisch gunstige reactie; de energie die hierbij vrijkomt
maakt het mogelijk dat in het metabolisme een thermodynamisch
ongunstige reactie toch kan plaatsvinden door deze te koppelen aan
de hydrolyse van een voldoend aantal moleculen ATP. In de
biosynthese worden daartoe niet-reactieve verbindingen met ATP
eerst omgezet in reactieve tussenproducten die daarna vlot de
vereiste reactie kunnen geven. Veel voorkomend is de omzetting
door ATP van een hydroxylgroep van een alcohol of een carbonzuur
in een fosfaatgroep, waarbij dan respectievelijk een
fosfaatester of een reactief acylfosfaat ontstaat. De
hydroxylgroep met zijn slechte vertrekkende eigenschappen wordt in
beide gevallen omgezet in een goede vertrekkende groep, namelijk
in het fosfaatanion.
Naast de energievoorziening helpen mitochondriën ook bij de
regulatie van concentraties calcium en andere elektrisch
geladen deeltjes in het cytoplasma. Ook recyclen zij- en breken
zij de energie in vetzuren en aminozuren af
.
Hoewel de mitochondriën al vanaf het jaar 1880 onderzocht worden,
duurde het vele jaren voordat wetenschappers het functioneren van
dit organel begrepen. Het proces waarbij de mitochondriën zuurstof
gebruiken om de chemische energie , die is opgeslagen in voedsel,
vrij te maken , wordt cellulaire ademhaling genoemd. ( cellular
respiration ).
In de vroege jaren 1900 werd ontdekt dat de biochemische reacties
van dit type ademhaling in twee hoofdgroepen vallen : het koolstof
traject , waarin suiker wordt afgebroken tot kooldioxide en
waterstof ; en het waterstof traject waarbij waterstof in stappen
wordt omgezet in zuurstof , waarbij water wordt gevormd en energie
vrijkomt.
In het waterstof traject passeren de elektronen van waterstof de
" elektronen transportketen " . Deze
elektronen transportketen bestaat uit 5
enzymcomplexen. Tijdens het transport van deze elektronen door de
elektronen transportketen, geven de elektronen
een deel van hun energie af. Deze energie wordt dan opgeslagen in
moleculen ATP. Aan het einde van de keten zijn voor elke molecuul
suiker, 38 moleculen ATP gevormd.
De overall energiebalans per glucose molecuul is:
| Aerobisch | Anaerobisch | |
| 2 ATP gebruikt in de glycolyse | - 2 ATP | - 2 ATP |
| 4 ATP gevormd in de glycolyse | + 4 ATP | + 4 ATP |
| 2 NADH2 gevormd in de glycolyse via elektronentransportketen | + 6 ATP | |
| 8 NADH2 gevormd in de citroenzuurcyclus via de e.t. | + 24 ATP | |
| 2 GTP in de citroenzuurcyclus | + 2 ATP | |
| 2 FADH2 in de citroenzuurcyclus via de elektronentransportketen | + 4 ATP | |
| Totaal : | 38 ATP | 2 ATP |
Soms wordt een totaal van 36 ATP aangehouden, omdat bekend is dat
in eukaryote cellen het gereduceerde NAD, dat gevormd is door de
glycolyse in het cytoplasma, actief getransporteerd moet worden
over de mitochondriële membraan om beschikbaar te komen voor de
elektronentransportketen.
Het actieve transport over het membraan kost 1 ATP voor elk NADH
dat getransporteerd wordt. Wanneer men zo wil redeneren , moet men
ook het actieve transport van andere moleculen in ogenschouw
nemen. ( pyruvaat ? , fosfaat ? , Mg+ etc ).
Het juiste netto resultaat van ATP is onbekend , maar moet worden
beschouwd als minder dan 36.
Mitochondriën zijn verbazingwekkend efficiënt bij de omzetting van
de chemische energie in suiker tot ATP. Een
verbrandingsmotor wordt al efficiënt beschouwd , als het 25
procent van de chemische energie die is opgeslagen in gasolie,
omzet in bewegingsenergie.
Mitochondriën echter, zetten 54 procent van de energie die is
opgeslagen in suiker, om in ATP.
Hoewel het al lang werd vermoed werd pas in 1988 bewezen dat
defecten in mitochondriële genen konden leiden tot erfelijke
ziekten. In 1988 toonde Douglas Wallace van de Emory University
aan dat de zeldzame ziekte "
Leber's hereditary optic neuropathy
" werd veroorzaakt door mutaties in het mitochondriële
DNA.
Mitochondriën oxideren korte- , midden - en grotendeels lange
keten vetzuren, terwijl
peroxisomen de
meeste zeer lang keten vetzuren en sommige lange keten vetzuren
oxideren.
Mitochondriën gebruiken een acyl-CoA dehydrogenase om acyl-CoA om
te zetten tot enoyl-CoA. Dit enzym transporteert de elektronen
naar FAD en vervolgens naar de
elektronentransportketen. Peroxisomen gebruiken
een acyl-CoA oxidase welke uiteindelijk de elektronen naar
zuurstof transporteert, waarbij waterstofperoxide wordt gevormd
die weer door catalase wordt omgezet in water en zuurstof. Verder
worden de mitochondriële en peroxisomale enzymen gecodeerd door
verschillende genen.
Het carnitine palmitoyltransferase systeem en de beta-oxidatie:
De hoofdbron voor de energie productie in de cel is de
mitochondriële beta-oxidatie van lang-keten vetzuren. Lang-keten
vetzuren kunnen niet direct de mitochondriële membranen passeren
en moeten eerst worden omgezet. Dit in tegenstelling tot de kort -
keten en middellange - keten vetzuren die door
diffusie de membranen kunnen passeren.
De oxidatie van lang-keten vetzuren in het mitochondrion
worden gekatalyseerd door de enzymen CPT I en CPT II . Samen met
acyl CoA synthetase en carnitine-acylcarnitine translocase
verzorgen zij het transport van vetzuren over de membranen van het
mitochondrion naar de mitochondriële matrix, waar deze vetzuren
uiteindelijk worden omgezet in energie in de vorm van
ATP.
Om de mitochondriële membranen te kunnen passeren , worden de
lang-keten vetzuren eerst geactiveerd door coënzym A en dan
verbonden met L-carnitine, een reactie die gekatalyseerd wordt
door het enzym carnitine palmitoyltransferase ( CPTase ).
Een deel van de CPTase activiteit ( CPT I ) is geassocieerd met de
buitenste mitochondriële membraan en katalyseert de vorming van
palmityl-carnitine. Palmityl-carnitine kan als carnitine ester de
buitenste membraan passeren.
Een andere activiteit van CPTase ( CPT II ) wordt geassocieerd met
de binnenste mitochondriële membraan . CPT II zet in het
mitochondrion palmityl-carnitine om in palmityl-CoA.
Tot slot initiëren CPT II en carnitine-acylcarnitine translocase
de mitochondriële oxidatie van lang-keten vetzuren.
Het Carnitine Palmitoyltransferase ( CPT ) systeem, heeft
betrekking op twee verschillende proteïnen, die gelokaliseerd zijn
op de buitenste - en binnenste mitochondriële
membranen.
CPT I wordt op de buitenste mitochondriële membraan gevonden en
CPT II wordt op de binnenste mitochondriële membraan gevonden.
CPT II wordt in alle cellen gevonden.
Van CPT I bestaan twee weefsel-specifieke isoformen, die L-CPT I
en M-CPT I worden genoemd.
L-CPT I wordt voornamelijk in de lever gevonden. M-CPT I wordt
voornamelijk in de spieren gevonden.
Het gen dat voor L-CPT I codeert, wordt CPT1A genoemd. Het
gen dat codeert voor M-CPT I , wordt CPT1B genoemd. Het gen
dat voor CPT II codeert, wordt CPT2 genoemd.
Het gen dat codeert voor CPT I is gelokaliseerd op chromosoom
11q13. Het gen dat codeert voor CPT II , is gelokaliseerd op
chromosoom 1p32.
De officiële naam voor het enzym carnitine palmitoyl transferase
is carnitine O-palmitoyltransferase.
EC nummer
2.3.1.21
Het enzym katalyseert de volgende reactie:
Palmitoyl-CoA + L-carnitine <=> CoA + L-palmitoylcarnitine
Stoornissen in dit systeem leiden tot de
carnitine palmitoyltransferase stoornissen.
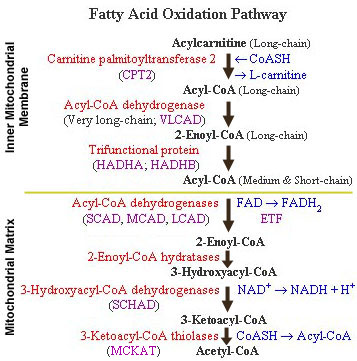
De mitochondriële β-oxidatie spiraal:
De beta-oxidatie: de afbraak (dehydrogenering ) vindt plaats
tussen de alfa en bèta koolstofatomen ( C2 en
C3 ) in een FAD gebonden reactie, die gekatalyseerd
wordt door acyl CoA dehydrogenase.
De producten van de beta-oxidatie zijn acetyl CoA,
FADH2, NADH en H+.
Acetyl CoA is acetyl coënzym A; FADH2 is de
gereduceerde vorm van flavine adenine dinucleotide en NADH is de
gereduceerde vorm van nicotinamide adenine dinucleotide.
De bètaoxidatie spiraal is een cyclus waarin acyl-CoA esters
steeds weer de cyclus doorlopen totdat ze volledig geoxideerd
zijn. Bij elke omgang in de cyclus wordt de ketenlengte met twee
koolstofatomen verkort .
Als voorbeeld kijken we eens naar Palmitoyl-CoA die een
ketenlengte van 16 koolstofatomen heeft.
De overall reactie is :
7 FAD + 7 NAD+ + 7 CoASH + H2O +
H(CH2CH2)7CH2CO-SCoA
> 8 CH3CO-SCoA + 7 FADH2 + 7 NADH + 7
H+
Palmitoyl-CoA maakt 7 omgangen in de bètaoxidatie spiraal door.
Bij elke omgang wordt de ketenlengte twee koolstofatomen korter,
waarna er uiteindelijk 8 delen met een ketenlengte van twee
koolstofatomen overblijven.
In de meeste weefsels zoals het hart en de spieren, worden deze 8
delen als acetyl-CoA in de
citroenzuur cyclus uiteindelijk omgezet in kooldioxide en water. In de lever
en de nieren wordt acetyl-CoA uit de β-oxidatie omgezet in
ketonen die daarna
voor de uiteindelijke oxidatie worden getransporteerd naar
bijvoorbeeld het brein en de spieren.
FADH2 , NADH en H+ worden geoxideerd in de
elektronen transportketen waarbij ATP wordt
gevormd.
Elke ronde van de ß-oxidatie-spiraal wordt gereguleerd door
een reeks van enzymen, waarvan sommige specificiteit
bezitten voor de ketenlengte van het acyl-CoA vetzuur.
Voor een verzadigd acyl-CoA vetzuur (zoals bijvoorbeeld
palmitoyl-CoA ( C16 )) worden vier
enzymstappen gevolgd, te weten die van:
De ketenlengte-specifieke acyl-CoA dehydrogenase enzymen zijn
flavoproteïnen; in het proces waarbij een dubbele binding gevormd
wordt tussen het a- en ß- koolstofatoom van een
acyl-CoA-vetzuur (en waarbij dus het corresponderende
enoyl-CoA-vetzuur ontstaat), worden de vrijgekomen elektronen
afgegeven aan ETF (elekton transfer proteïn) of ETF-dehydrogenase;
dit complex legt de link tussen ß-oxidatie en ademhalingsketen.
De tweede reaktie in de ß-oxidatie wordt gekatalyseerd door
enoyl-CoA hydratase (crotonase). Het enoyl-CoA-vetzuur
wordt hierbij gehydrateerd tot een L-ß-hydroxyacyl-CoA-vetzuur.
Vervolgens katalyseert ß-hydroxyacyl-CoA dehydrogenase (bestaande
uit 1 of meer enzymen) in een NAD+-afhankelijke
reaktie, oxidatie van de hydroxy-groep tot een ketogroep.
Bij de laatste reaktie, die gekatalyseerd wordt door
ß-ketoacyl-CoA thiolase (ook weer bestaand uit 1 of meer
enzymen) wordt in aanwezigheid van gereduceerd CoA, de a-ß binding
verbroken.
De twee produkten die bij deze reaktie
ontstaan zijn acetyl-CoA, en een acyl-CoA vetzuur dat 2
koolstofatomen korter is dan het oorspronkelijke substraat.
Zolang acyl-CoA-vetzuren een acetyl-CoA fragment af kunnen
splitsen, kunnen ze de ß-oxidatie blijven doorlopen.
Er bestaan drie acyl-CoA dehydrogenase enzymen, die ieder een
verschillende ketenlengte-specificiteit bezitten; hoewel er
overlap bestaat tussen deze enzymen, kan gezegd worden dat in de
lever (V)LCAD ((Very) Long Chain Acyl-CoA Dehydrogenase) de eerste
reaktie in de ß-oxidatie katalyseert voor acyl-CoA-vetzuren
met een ketenlengte van 18 tot 12 koolstofatomen.
MCAD (Medium Chain Acyl-CoA Dehydrogenase ) herkent
acyl-CoA-vetzuren met een ketenlengte variërend van 14 tot 4
koolstofatomen, en SCAD ( Short Chain Acyl-CoA Dehydrogenase )
herkent alleen vetzuurketens van 4en 6 koolstofatomen.
Vetzuren met een oneven ketenlengte worden ook in de bèta-oxidatie
spiraal afgebroken tot er een molecuul met 3 koolstofatomen
overblijft. Dit molecuul ( propionyl CoA ) kan niet verder in de
bèta-oxidatie spiraal worden afgebroken, maar wordt omgezet tot
succinyl CoA ( barnsteenzuur Coënzym A ) en wordt vervolgens
verder omgezet in de
citroenzuurcyclus.
Onverzadigde vetzuren:
Onverzadigde vetzuren zijn vetzuren die een of meer dubbele
bindingen in hun alkyl-keten hebben.
Meervoudig onverzadigde vetzuren met twee dubbele bindingen
hebben normaal gesproken een dubbele binding op C9 - C10 in de
alkyl-keten en een dubbele binding 3 C verder in de keten op
C12- C13.
De bètaoxidatie van deze vetzuren wordt doorlopen tot het punt
waarop de normale enzymen van de bètaoxidatie een dubbele
binding tegen komen. De enzymen van de bètaoxidatie zijn niet in
staat om op substraten te reageren die een dubbele binding
hebben tussen de bèta en gamma C-atomen.
Om dit probleem op te lossen, verplaatst het enzym enoyl-CoA
isomerase de dubbele binding een C atoom verder naar het einde
van de alkyl-keten, waarna de bètaoxidatie weer een ronde verder
( en een deel van de tweede ) kan verlopen tot de volgende
dubbele binding wordt aangetroffen waarna het enzym 2,4
dienoyl-CoA reductase het vetzuur reduceert waarbij een
trans-3-dubbele binding ontstaat. Het enzym 3,2 enoyl-CoA
isomerase verplaatst deze dubbele binding verder naar het einde
van de vetzuurketen, waar het vetzuur als substraat voor de
normale bètaoxidatie enzymen kan dienen.
Historie:
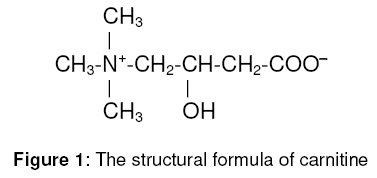
Carnitine ( 3-hydroxy-4-trimethyl aminoboterzuur :
C7H15NO3 ) is een kleine,
water oplosbare, quaternaire stikstof bevattende stof . Het wordt
in het lichaam in de lever, het brein en de nieren gesynthetiseerd
uit lysine en methionine ( met cofactoren zoals ascorbinezuur,
niacine , vitamine B6 en gereduceerd ijzer ).
Carnitine komt zowel in de L-vorm als in de D-vorm voor.
De L- vorm ( L-carnitine ) is de biologische actieve vorm.
Hoewel de mens in staat is carnitine uit lysine en methionine te
synthetiseren, moet tweederde van de benodigde carnitine uit het
voedsel gehaald worden.
Carnitine komt met name in rundvlees, schapenvlees en lamsvlees voor. Melk en kaas bevatten wat minder carnitine, terwijl fruit en groenten maar minieme hoeveelheden carnitine bevatten.
Carnitine is geen aminozuur , maar een vitamine-achtige
stof. Carnitine wordt niet afgebroken , maar wordt in de
urine uitgescheiden als vrije carnitine en veresterde vormen van
carnitine.
Carnitine functioneert in een driedelig enzymcomplex ( carnitine
acyltransferase I, carnitine translocase, en carnitine
acyltransferase II ) , die verantwoordelijk is voor het transport
van lange keten vetzuren over de binnenste mitochondriële
membraan.
Naast bovengenoemde sleutelfunctie speelt L-carnitine nog een rol
bij enkele andere metabole functies. Carnitine verbetert de
metabole flux in de
citroenzuurcyclus
en voorkomt de stapeling van lange keten Acyl CoA. Verder fungeert
L-carnitine als drager voor acyl-deeltjes ( verkort in de
peroxisomale beta-oxidatie ) van het peroxisoom naar het
mitochondrion, waar deze deeltjes verder geoxideerd kunnen
worden.
Carnitine is dus direct of indirect betrokken bij verschillende
metabole paden en de beschikbaarheid van carnitine is een
belangrijke factor die niet alleen de oxidatie van vetzuren en
ketonlichamen regelt, maar ook die van glucose en andere
aminozuren..
De Synthese van Carnitine in het lichaam:
De elektronen transportketen.( de ademhalingsketen).
Bij de ademhaling wordt zuurstof opgenomen dat via de longen en
de bloedbaan getransporteerd wordt naar de mitochondriën in
de cel.
De functie van zuurstof is de verschillende
brandstofmoleculen te oxideren, waarbij energie vrijkomt
die voor tal van processen benut kan worden. Bij deze
oxidatiereacties worden uiteindelijk elektronen van een
substraat overgedragen op zuurstof. In biologische oxidaties
vindt deze elektronenoverdracht echter niet rechtstreeks plaats,
maar verloopt via een vaste serie reacties die bekend staat als
de elektronen transportketen of de ademhalingsketen.
Aan de elektronen transportketen neemt een aantal verbindingen deel die ieder voor zich goed een reversibele redoxreactie kunnen ondergaan. Iedere schakel in de keten reduceert een volgende schakel en wordt daarbij zelf geoxideerd.
Het NAD+/NADH-redoxkoppel heeft de meest negatieve reductie potentiaal en kan dus het gemakkelijkst elektronen doorgeven aan andere systemen.
Het elektronenverloop in de elektronen transportketen volgt de thermodynamisch strikt logische route van NADH ( twee elektronenoverdracht ) via de flavinen FMN of FAD naar coënzym Q. Dit coënzym draagt vervolgens de elektronen via vijf cytochromen ( b,c1,c,a en a3; één-elektronprocessen ) over op zuurstof (vier-elektronenproces).
Coënzym Q is een knooppunt in de elektronen transportketen, want het sluist de elektronen van twee verschillende redoxpaden door naar zuurstof via het cytochromensysteem.
Coënzym Q is de enige verbinding in de elektronen transportketen die niet sterk gebonden zit aan een eiwit.
De isopreenketen aan coënzym Q maakt de verbinding zeer apolair, waardoor deze zich gemakkelijk in de apolaire omgeving van de mitochondriën kan bewegen en zodoende een mobiele schakel vormt tussen flavoproteïnen en cytochromen.
Bij het elektronentransport vindt een geleidelijke verlaging van energie plaats. Een deel van de energie wordt opgeslagen in de vorm van ATP doordat het elektronentransport in de mitochondriën gekoppeld is aan de productie van ATP uit ADP en fosfaat.( ATP-adenosine tri-fosfaat;ADP-adenosine di-fosfaat ).
Dit proces wordt oxidatieve fosforylering genoemd.
Reacties:
NAD+ + H+ + 2 e Û NADH
riboflavine + 2H+ + 2 e Û dihydroriboflavine
coënzym Q10 + 2H+ + 2 e Û dihydrocoënzym Q10
cytochroom c (Fe3+)+ e Û cytochroom c (Fe2+)
cytochroom a3 (Fe3+) + e Û cytochroom a3 (Fe2+)
O2 + 4H+ + 4 e Û
2H2O
De oxidatieve fosforylering ( oxidative phosphorylation or OXPHOS).
Oxidatieve fosforylering :
Oxidatie is het proces waarbij elektronen worden afgestaan en fosforylering is het proces waarbij een fosfaatgroep wordt opgenomen.
Oxidatie vindt plaats in de volgende omkeerbare reactie :
NADH + ubiquinone <=> NAD+ + ubiquinol
Fosforylering vindt plaats in de volgende omkeerbare reactie :
ATP + H2O <=> ADP + fosfaat
De oxidatieve fosforylering is verantwoordelijk voor de meeste ATP (adenosine tri-fosfaat) die de cellen nodig hebben. ATP is de bron van cel-energie in alle levende wezens en niet alleen noodzakelijk om te leven, maar voor praktisch alle lichaamsfuncties, door de elektrische energie die daarvoor nodig is.
Het OXPHOS pad omvat meer dan 100 polypeptiden wiens genen of in het DNA van de celkern– of in het DNA van het mitochondrion gelokaliseerd zijn. De expressie van deze genen en de samenstelling van de vijf OXPHOS enzym complexen ( I t/m V) is een zeer geordend en gecoördineerd proces.
Oxphos bestaat uit vijf enzym complexen. Deze enzymen zijn gelokaliseerd in het mitochondriële binnenste membraan en worden als volgt benoemd:
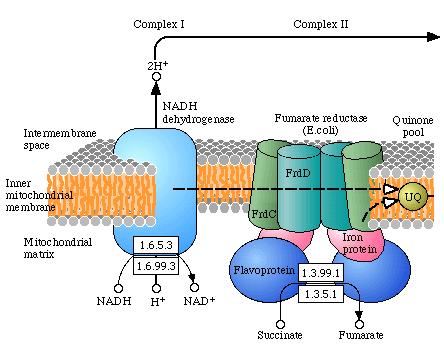
Complex I : NADH: ubiquinone oxidoreductase ( EC 1.6.5.3 )
Complex II : succinate:ubiquinone oxidoreductase ( EC 1.3.5.1 )
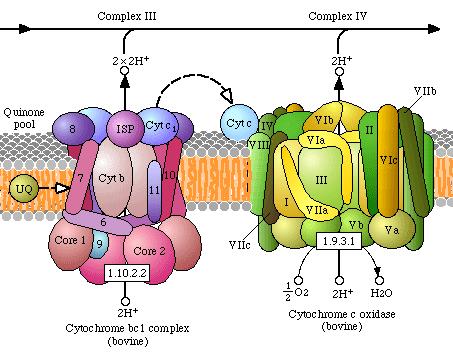
Complex III : ubiquinol: ferrocytochroom c oxidoreductase ( EC 1.10.2.2 )
Complex IV: ferrocytochroom c : oxygen oxidoreductase of cytochroom c oxidase ( EC 1.9.3.1 )
Compex V : H(+)-transporting two-sector ATPase ( EC 3.6.3.14 ) Formerly EC 3.6.1.34
Complex I en II collecteren de elektronen uit verschillende bronnen en geven ze af aan ubiquinone (coënzym Q10).
De elektronen bewegen zich dan door de complexen III en IV en reageren dan uiteindelijk met zuurstof.
Het is nu duidelijk dat ziekten kunnen ontstaan door mutaties in de genen van de celkern of in genen van het mitochondrion en er kunnen zelfs ziekten ontstaan door de fouten in systemen die de interactie tussen deze twee coördineren.
Om deze redenen hebben OXPHOS ziekten een complexe reeks van erfelijkheid en hebben een breed spectrum aan klinische presentaties.
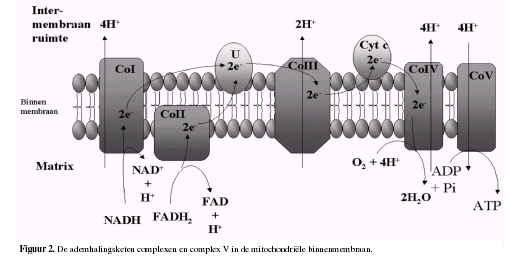
Complex I, III en IV overspannen de binnenste
mitochondriële membraan en zijn protonen pompen.
Complex II bevindt zich tussen complex I ( NADH: ubiquinone
oxidoreductase ) en Complex III ( ubiquinol: ferrocytochroom c
oxidoreductase ).
Complex II overspant de binnenste mitochondriële membraan niet,
maar bevindt zich aan de matrix-zijde van de binnenste
mitochondriële membraan en is het ingangspunt van elektronen
afkomstig van FADH2. Complex I is het ingangspunt
voor elektronen afkomstig van NADH.
Alle 4 genoemde enzymen bevatten prosthetische groepen die de
eigenlijke elektronendragers in de enzymen zijn.
In elk complex en ook van complex naar complex zijn
verschillende groepen betrokken bij de eigenlijke elektronen
overdracht. Deze groepen kunnen worden onderscheiden in:
ijzer-zwavel proteïnen; haem; koper en flavinen. Allen dienen
als elektronendrager, echter elk enzymcomplex wordt geassocieerd
met bepaalde prostethetische groepen:
Complex I: FMN en Fe-S
Complex II: FAD en Fe-S
Complex III: Heme b-562, Heme b-566, Heme c1,
Fe-S
Complex IV: Heme a, Heme a3, CuA,
CuB


Copyright 1997. Thomas M. Terry, The University of
Connecticut.
Het schema hierboven illustreert een mitochondrion. Bekijk in de animatie hoe NADH H+ ionen en elektronen verplaatst in de elektronen transportketen.
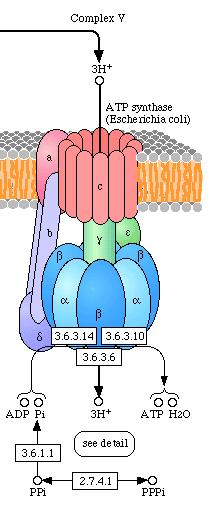
Stap 1: Een Proton gradiënt wordt opgebouwd doordat NADH elektronen in de elektrontransportketen brengt..
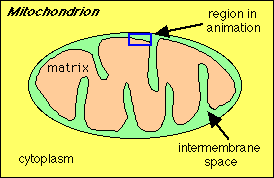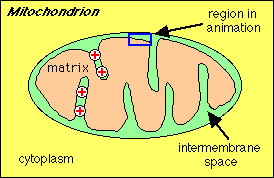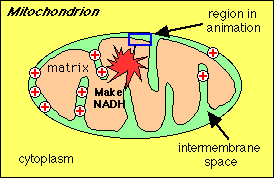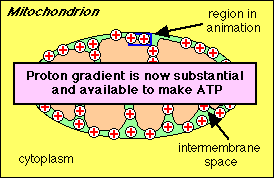
Stap 2: Protonen (aangeduid door + lading) komen terug in de mitochondriële matrix door kanalen in het ATP synthase enzym complex. ATP wordt gesynthetiseerd uit ADP en fosfaat (Pi)
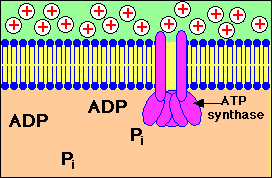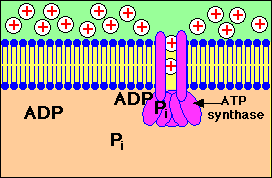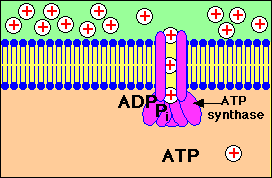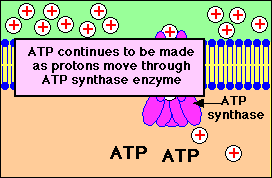
Bekijk in bovenstaande animatie hoe er een opbouw plaatsvindt
van H+ ionen in de ruimte tussen de binnenste en
buitenste mitochondriele membraam telkens wanneer NADH
geproduceerd wordt uit oxidatieve reacties.
protonen komen de matrix weer binnen door middel van het ATP
synthase complex waarna ATP geproduceerd wordt.
Protonen worden over het membraan getransporteerd; dat wil
zeggen: vanuit de matrix naar de ruimte tussen de binnenste en
de buitenste membraan.
Terwijl NADH steeds meer H+ en elektronen in de
elektronen transportketen overbrengt, zal de proton gradiënt
toenemen, met een opbouw van H+ buiten de binnenste
mitochondriële membraan en OH- binnen de binnenste
mitochondriele membraan.
Er ontstaat een proton gradiënt.
ATP synthase
is een groot proteïne complex met een proton kanaal
die de herintreding van protonen toestaat.
ATP synthese wordt aangedreven door de resulterende stroom van
protonen die door het membraan stromen:
ADP + Pi ---> ATP
Animaties:
ATP synthase
http://www.stolaf.edu/people/giannini/flashanimat/metabolism/atpsyn1.swf
This short animation illustrates the production of ATP by ATP
synthase.
ATP synthase mechanism
http://www.stolaf.edu/people/giannini/flashanimat/metabolism/atpsyn2.swf
This quick animation provides a look at the action of ATP
synthase.
Mitochondrial electron transport
http://www.stolaf.edu/people/giannini/flashanimat/metabolism/mido%20e%20transport.swf
Click here for a brief but clear animation demonstrating the
electron transport chain.
Algemeen.
De genen zijn een deel van de chromosomen die op hun beurt
gelokaliseerd zijn in de nucleus van elke cel.
De chromosomen zijn opgebouwd uit een verbinding die DNA wordt
genoemd.
De mitochondriën, die zich buiten de nucleus in het cytoplasma
van de cel bevinden, zijn een andere plaats waar DNA wordt
gevonden.
Dus ook de mitochondriën bevatten genen; alhoewel het
mitochondriële DNA één lange streng genen is; en niet is
samengesteld uit chromosomen.
De hoeveelheden mitochondriën in elke cel kan variëren van 1 tot
duizenden.
Al deze mitochondriën; en dus ook het DNA in de mitochondriën,
stammen af van een klein aantal mitochondriën die aanwezig waren
in de originele eicel op het moment van de conceptie. Het
sperma draagt niet bij aan de mitochondriën van de baby. Dus de
mitochondriën worden alleen geërfd van de moeder. (Zeer zeldzaam
voorkomend worden mitochondriën
van de vader
geërfd ).
Een abnormaliteit in één van de genen in de mitochondriën kan
daardoor worden doorgegeven in de eicellen van de
moeder. Daar de meeste eicellen van de moeder dezelfde
mitochondriële mutatie bevatten, is het risico op een
volgend kind met een mitochondriëleaandoening hoog.
Dit patroon van erfelijkheid wordt
maternale overerving genoemd. (maternaal = van de
moeder).
Veel aangeboren mitochondriële aandoeningen worden nucleus
defecten genoemd (Een fout in een gen in de kern van de cel);
met een overerving van: ofde vader, of de moeder of van zowel de
vader als de moeder. Dit patroon wordt
autosomaalrecessief erfelijk genoemd. Deze kunnen
dominant of recessief zijn. In dit geval is de kans op een
aandoening in het volgend kind 25%. Verder is er nog een
X-gebonden overerving; deze heeft betrekking op defecten
van het X-chromosoom. Nog een andere mogelijkheid is de kans dat
er tijdens de bevruchting een mutatie is opgetreden in een gen
en dat dit kind de eerste in een familie is die ziek
wordt.
De effecten van mutaties in de mitochondriële genen.
De eicel bevat veel mitochondriën en elk mitochondrion bevat
verschillende kopieën van de mitochondriële genen.
Wanneer een specifiek mitochondrieel gen in elk mitochondrion in
de eicel gemuteerd zou zijn en daarom de verkeerde
boodschap uit zou zenden,dan zou de breuk in de energie
productie zo ernstig zijn dat de vroege embryo dit niet zou
overleven.
( Anders gezegd: pas wanneer een
aanvankelijk laag percentage gemuteerde mtDNAs na een zeker aantal
celdelingen een bepaalde drempel overschrijdt, leidt dit (soms
vrij plotseling) tot problemen.
Omdat het aantal mtDNA kopieën per mitochondrion en per cel,
alsmede de behoefte aan oxidatieve fosforylering per weefsel
verschilt, heeft elk weefsel zijn eigen typische
drempelwaarde.
)
Dus; een persoon die geboren is met een erfelijke mitochondriële
ziekte moet twee typen mitochondriën van zijn of haar
moeder hebben geërfd: enkele die de correcte kopieën van het gen
bevatten en enkele die de gemuteerde en ziekmakende kopieën
van het gen bevatten.
In tegenstelling tot de meeste genetische ziekten waarbij de
mutatie in alle cellen van het lichaam wordt gezien, worden
mutaties van het mitochondriële DNA niet in alle cellen van het
lichaam gezien.
Mitochondriën worden willekeurig verdeeld in de eicel wanneer
deze gevormd wordt in het ovarium. Daarom zal de samenstelling
van de mitochondriën in elke eicel variëren van meest normaal
tot meest abnormaal.
Daarom zullen alle kinderen van deze moeder enige abnormale
genen erven, maar alleen symptomen ontwikkelen
wanneer de mitochondriën met het gemuteerde gen een zeker
kritisch niveau hebben bereikt met de interferentie van de
energie productie in het lichaam.
Het is dus niet te voorspellen in welke mate iemand ziek zal
worden en welke organen daar eventueelbetrokken bij
zijn.
Evenmin is het te voorspellen wat het verloop van de ziekte zal
zijn over een bepaalde tijd, daar men weinig
zal kunnen zeggen over de verdeling en de hoeveelheid van
de mitochondriën met het gemuteerde gen in het lichaam. De
gemuteerde mitochondriën kunnen gelokaliseerd zijn in de
skeletspieren, of in het hart ,of de nieren, of in
het brein of in de lever, enzovoort. Of in combinaties bij
bovenstaande , of in alle organen.
Dit maakt de mitochondriële aandoeningen zo complex.
Tot de mitochondriële ziekten behoren o.a. :
Stoornissen in de Carnitine cyclus:
defecten van de bèta-oxidatie spiraal:
Meer informatie: VDAC Web Page
Meer informatie: Structure of the voltage dependent, ion selective channel (VDAC)
Voltage-Dependent Anion Channel 1; VDAC1
OMIM:
Voltage Dependent Anion Channel Deficiency ( VDAC
Deficiency )
Voltage-Dependent Anion Channel 2; VDAC2
OMIM:
Voltage-Dependent Anion Channel 2; VDAC2
( indeling van de ziektes naar de verschillende complexen )
( Indeling van de ziektes als verschillende syndromen )
Leber hereditary optic neuroretinopathy (LHON).
OMIM:
535000
OMIM:
Clinical Synopsis
GeneReviews:
Leber Hereditary Optic Neuropathy
Who named it?:
Theodor Karl Gustav von Leber
|
|

|
Hoofdmenu |
|
|
|
|
|
|
 Mitochondrion
Mitochondrion